- Home
- Resource
- Disease Diagnosis
- Metabolic Diseases
- Beyond Elevated Homocysteine: A Strategic Guide to Differential Diagnosis of Homocystinuria
- Home
- IVD
- By Technology Types
- By Diseases Types
- By Product Types
- Research
- Resource
- Distributors
- Company

Homocystinuria represents a complex group of inherited metabolic disorders where accurate and early differential diagnosis is critical for implementing targeted interventions to prevent severe thrombotic, neurological, and ocular complications. This resource provides a strategic overview of the diagnostic pathway for homocystinuria, emphasizing the integration of key biomarkers to distinguish between subtypes such as cystathionine beta-synthase (CBS) deficiency and remethylation defects.
Homocystinuria is a group of inherited metabolic disorders characterized by the accumulation of homocysteine and its metabolites due to defects in the transsulfuration or remethylation pathways, most commonly caused by deficiency of cystathionine beta-synthase (CBS) or impairments in cobalamin (B12) metabolism. This accumulation leads to multisystemic complications including thromboembolism, lens dislocation, skeletal abnormalities, and developmental delay. Early and accurate biochemical and genetic diagnosis is critical to initiate targeted interventions that can prevent severe clinical outcomes and improve long-term prognosis.
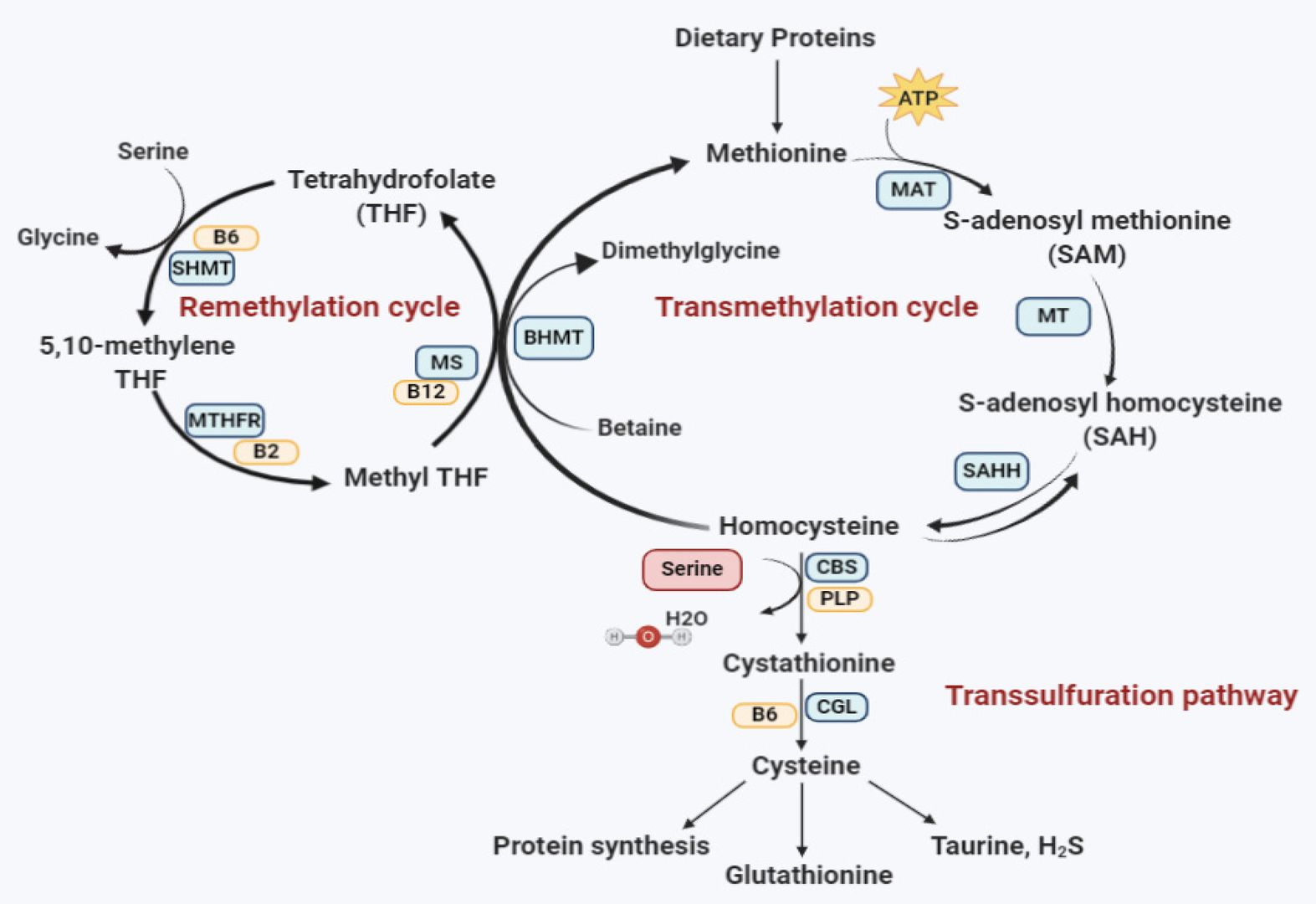 Fig.1 Homocysteine metabolism pathway. (Al-Sadeq D W, Nasrallah G K., 2020)
Fig.1 Homocysteine metabolism pathway. (Al-Sadeq D W, Nasrallah G K., 2020)
The diagnosis and differential diagnosis of homocystinuria rely on a precise interpretation of a panel of key biochemical biomarkers measured in blood and urine. These markers not only confirm the presence of the disease but are essential for distinguishing between its various genetic causes, which is critical for initiating the correct treatment strategy.
The diagnostic pathway for homocystinuria is a systematic, multi-stage process designed to efficiently confirm hyperhomocystinemia, differentiate between its numerous genetic causes, and guide definitive testing and treatment. This strategy is crucial because management varies significantly depending on the underlying defect.
Initial Screening and Confirmation
The pathway begins with measuring plasma total homocysteine (tHcy) to confirm hyperhomocystinemia. This critical first step distinguishes potential genetic disorders from other causes and triggers further specialized testing.

Core Differential Diagnosis
The process then branches based on methionine levels from plasma amino acid analysis. Elevated methionine suggests classic CBS deficiency, while low/normal methionine points toward a remethylation pathway defect, guiding subsequent testing strategies.

Remethylation Defect Subclassification
For cases with low methionine, methylmalonic acid (MMA) and vitamin B12 levels are analyzed. Elevated MMA indicates a cobalamin metabolism disorder (e.g., cblC), while normal MMA suggests a pure remethylation defect (e.g., MTHFR deficiency).
Genetic Confirmation
The pathway concludes with targeted genetic testing of genes corresponding to the biochemical profile (e.g., CBS, MMACHC, MTHFR). This provides definitive diagnosis, enables family studies, and informs precise long-term management strategies.

Navigating the diagnosis of homocystinuria involves overcoming significant complexities, including the variable phenotypic expression of disorders, the potential for nutritional deficiencies (e.g., B12 deficiency) to mimic genetic causes, and the existence of mild or late-onset forms that evade classic biochemical patterns. Additionally, interpreting genetic variants of uncertain significance (VUS) and differentiating between closely related remethylation defects require integrating multiple biomarkers—such as homocysteine, methionine, methylmalonic acid (MMA), and vitamins—within the clinical context to avoid misdiagnosis and ensure accurate classification for targeted treatment.
The future of homocystinuria diagnostics is advancing toward integrated multi-omics approaches, combining high-throughput genetic screening (e.g., next-generation sequencing panels) with refined biochemical profiling via tandem mass spectrometry (MS/MS) and AI-driven data interpretation. These innovations aim to enhance newborn screening precision, accelerate differential diagnosis between subtypes (e.g., CBS deficiency vs. remethylation defects), and enable earlier detection of atypical cases. Emerging technologies like liquid chromatography-high-resolution mass spectrometry (LC-HRMS) will further streamline simultaneous quantification of homocysteine, methylmalonic acid, and related metabolites, paving the way for personalized therapeutic strategies and improved long-term patient outcomes.
As a leading provider of IVD solutions for metabolic diseases, Alta DiagnoTech offers a comprehensive portfolio of homocystinuria diagnostics, including immunoassays for total homocysteine and vitamins, LC-MS/MS solutions for amino acids and methylmalonic acid, and genetic testing panels for subtype confirmation. If you have related needs, please feel free to contact us for more information or product support.
Reference
This article is for research use only. Do not use in any diagnostic or therapeutic application.
|
There is no product in your cart. |
Copyright © 2026 Alta DiagnoTech. All rights reserved.