- Home
- Resource
- Disease Diagnosis
- Metabolic Diseases
- Optimizing the Diagnosis of Cystinosis: Integrating Biochemical and Genetic Testing
- Home
- IVD
- By Technology Types
- By Diseases Types
- By Product Types
- Research
- Resource
- Distributors
- Company

Cystinosis is a rare, inherited lysosomal storage disorder caused by mutations in the CTNS gene, leading to destructive cystine crystal accumulation across multiple organs. Timely and accurate diagnosis is critical to prevent irreversible renal failure and systemic complications. This resource details the optimized diagnostic pathway for cystinosis, highlighting the essential integration of biochemical quantification of leukocyte cystine with molecular genetic confirmation of CTNS mutations.
Cystinosis is a rare, autosomal recessive lysosomal storage disorder caused by mutations in the CTNS gene, which encodes the cystine transporter protein cystinosin. This defect leads to the accumulation of cystine crystals within lysosomes, resulting in progressive cellular damage across multiple organs. Without early diagnosis and treatment, the disease progresses to end-stage renal failure and extra-renal complications. Timely biochemical and genetic diagnosis is critical to initiate life-saving therapies like cysteamine and improve long-term outcomes.
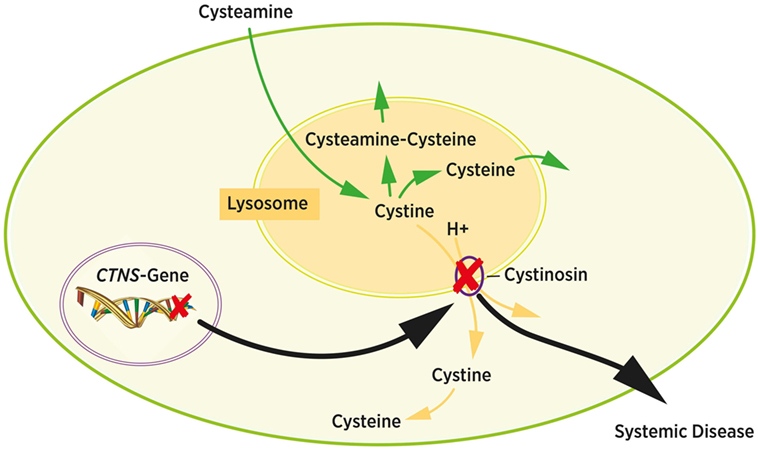 Fig.1 Intracellular cystine metabolism. (Bäumner S, Weber L T., 2018)
Fig.1 Intracellular cystine metabolism. (Bäumner S, Weber L T., 2018)
Clinical suspicion for cystinosis arises primarily in infants and young children presenting with failure to thrive, polyuria, polydipsia, and dehydration due to renal Fanconi syndrome—a hallmark feature characterized by generalized tubular dysfunction manifesting as glycosuria, phosphaturia, and metabolic acidosis. Additional key indicators include photophobia (from corneal cystine crystal deposition), hypophosphatemic rickets, and unexplained hypothyroidism. In older patients or atypical cases, corneal crystals on slit-lamp examination or progressive glomerular dysfunction without obvious cause may prompt diagnostic evaluation. Early recognition of these signs is crucial to initiate targeted diagnostic testing and prevent end-organ damage.
The quantitative measurement of cystine in white blood cells (WBCs) is the unequivocal biochemical gold standard for diagnosing cystinosis. This test directly quantifies the pathological hallmark of the disease—the accumulation of cystine within lysosomes due to a defective cystinosin transporter. It provides functional evidence of the metabolic blockade, offering critical specificity to distinguish cystinosis from other disorders with similar clinical presentations, such as Fanconi syndrome.

Methodology
Leukocyte cystine measurement utilizes highly specialized techniques beginning with isolation of a pure white blood cell pellet from fresh whole blood. The sample undergoes protein precipitation and derivatization before quantification through advanced analytical platforms such as high-performance liquid chromatography (HPLC) or liquid chromatography-tandem mass spectrometry (LC-MS/MS). These methods provide the sensitivity and precision required to accurately measure intracellular cystine accumulation.

Interpretation
Diagnostic interpretation relies on established threshold values, with results reported in nmol half-cystine/mg protein. Levels below 0.2 nmol/mg are considered normal, while consistent measurements above this threshold confirm cystinosis diagnosis. For patients undergoing treatment, therapeutic monitoring aims to maintain cystine levels below 1.0 nmol/mg protein to ensure effective disease management and slowed progression.
Role in Optimization
This assay optimizes diagnostics by serving as a specific triage tool that directly confirms the biochemical defect, efficiently guiding subsequent genetic testing. It enables personalized treatment by providing quantitative data for cysteamine therapy titration and enhances variant interpretation by offering functional context for genetic findings. The integration of biochemical and genetic testing creates a streamlined, accurate diagnostic pathway that improves patient outcomes.
Genetic confirmation represents the definitive diagnostic step for cystinosis, directly identifying the underlying molecular defect in the CTNS gene (located on chromosome 17p13.2) that encodes the lysosomal cystine transporter cystinosin. This approach is critical for clarifying ambiguous cases, informing genetic counseling, and completing the diagnostic workflow with unequivocal precision.
Detection Methods
The future of cystinosis diagnostics is advancing toward non-invasive methods and integrated multi-omics approaches. Newborn screening via tandem mass spectrometry (MS/MS) of dried blood spots is being refined for earlier detection, while next-generation sequencing (NGS) panels improve genetic variant interpretation. Artificial intelligence may soon assist in predicting disease progression from biochemical and genetic data, enabling personalized monitoring and therapy optimization.
As a professional provider of in vitro diagnostic (IVD) solutions, Alta DiagnoTech offers leukocyte cystine quantification kits, CTNS gene Sanger sequencing reagents, and NGS-based mutation detection panels to promote accurate and efficient diagnosis of cystinosis. If you have related needs, please feel free to contact us for more information or product support.
Reference
This article is for research use only. Do not use in any diagnostic or therapeutic application.
|
There is no product in your cart. |
Copyright © 2026 Alta DiagnoTech. All rights reserved.