- Home
- Resource
- Explore & Learn
- The New Landscape of Laboratory-Developed Tests: A Regulatory Overview for Anatomic Pathology
- Home
- IVD
- By Technology Types
- By Diseases Types
- By Product Types
- Research
- Resource
- Distributors
- Company

Laboratory-developed tests (LDTs) have long been a critical component of personalized medical care, particularly in the field of anatomic pathology. Historically, LDTs were developed and used by clinical laboratories without specific regulatory oversight. The FDA’s authority over medical devices, including in vitro diagnostics (IVDs), was established by the Medical Device Amendments of 1976. However, LDTs were not explicitly mentioned in these amendments or in the subsequent Clinical Laboratory Improvement Amendments of 1988 (CLIA). This regulatory ambiguity led to a period where LDTs operated under a policy of enforcement discretion, allowing them to be used without the same level of scrutiny as commercial IVDs.
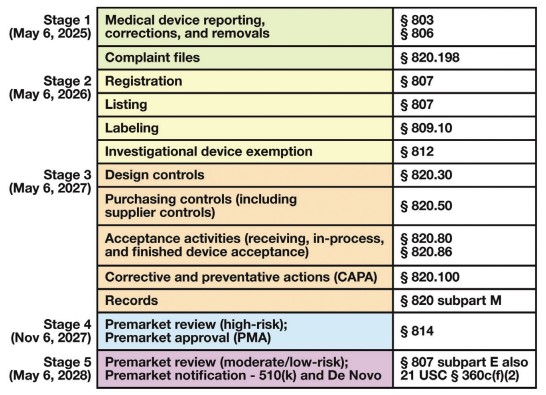 Fig.1 LDT Final Rule timeline of requirements. (Genzen J. R., et al., 2025)
Fig.1 LDT Final Rule timeline of requirements. (Genzen J. R., et al., 2025)
In 2010, the FDA announced its intention to regulate LDTs as IVDs, signaling a significant shift from its previous stance. This move was met with both support and criticism from various stakeholders in the medical community. The FDA’s proposed regulatory framework in 2014 generated substantial debate, highlighting the complexities and challenges of balancing innovation with patient safety.
After years of deliberation and public commentary, the FDA published its Final Rule on LDTs on May 6, 2024. This rule represents a culmination of efforts to clarify and enforce regulatory oversight over LDTs. The Final Rule was developed through a notice and comment rulemaking process, which included a 60-day public comment period. Despite requests for an extension, the FDA proceeded with its accelerated timeline, aiming to minimize congressional review under the Congressional Review Act.
The Final Rule essentially expands the definition of “in vitro diagnostic products” to include LDTs, asserting that the existing regulatory framework for commercial IVDs now applies to all LDTs. The FDA has ended its policy of general enforcement discretion over LDTs but has introduced targeted enforcement discretion for certain types of LDTs, such as those used in Veterans Health Administration settings, forensic testing, and specific unmet needs.

Enforcement Discretion Policies
The FDA’s targeted enforcement discretion includes exemptions for “1976-type LDTs,” which are manual tests using components legally marketed for clinical use within a single CLIA high-complexity laboratory. However, this exemption does not extend to tests incorporating automation, software, or research use only (RUO) reagents. The FDA also offers partial enforcement discretion for LDTs that were marketed before the Final Rule’s publication, tests for rare conditions, and tests approved by the New York State Clinical Laboratory Evaluation Program (NYS CLEP).
The FDA’s categorization of manual immunohistochemistry (IHC) and in situ hybridization (ISH) tests as 1976-type LDTs has significant implications for anatomic pathology. These tests, which are fundamental to diagnosing and classifying tumors, are now subject to regulatory scrutiny. However, the FDA’s decision to exclude automated testing from this category means that most modern clinical laboratories will not qualify for this exemption.
The unmet needs provision in the Final Rule aims to address the availability of tests for rare disorders. However, this provision comes with significant limitations, such as restrictions on outreach testing and the requirement that tests be ordered by healthcare practitioners within the same healthcare system. This limits the applicability of the provision in reference laboratory settings, which are crucial for diagnosing rare conditions.
The FDA expects compliance with quality system (QS) and premarket review requirements for LDTs when significant modifications are made. This includes changes in the indications for use, operating principles, or the introduction of new technologies. For anatomic pathology, this means that even routine optimizations of IHC protocols may require additional regulatory steps.
Understanding the FDA’s classification of medical devices is crucial for compliance with the Final Rule. The FDA categorizes devices into three classes based on risk: Class I (low risk), Class II (moderate risk), and Class III (high risk). Each class has specific regulatory requirements, ranging from general controls for Class I devices to premarket approval (PMA) for Class III devices.
General purpose reagents, such as those used for tissue processing and staining, are classified as Class I devices and are subject to general controls. IHC kits and reagents, which are vital for anatomic pathology, are categorized based on their intended use and risk level. For example, Class I IHC reagents provide adjunctive diagnostic information, while Class II and III reagents offer prognostic or predictive data.
The rise of digital pathology and AI-based tools has introduced new regulatory challenges. The FDA has issued guidance documents and approved several whole-slide imaging systems for digital pathology. These systems and AI algorithms are classified as Class II devices, subject to special controls and premarket review.
The LDT Final Rule outlines a five-stage phase-out policy over four years, with specific deadlines for compliance. Pathologists and laboratory directors must be aware of these deadlines and the associated requirements, such as medical device reporting (MDR) and complaint file handling, which are due by May 6, 2025.
Compliance with the Final Rule requires a thorough understanding of device classifications and product codes. Clinical laboratories must ensure that their LDTs are correctly classified and listed with the FDA. This process involves selecting appropriate product codes and adhering to applicable regulatory requirements.
The FDA’s 2024 Final Rule on LDTs marks a significant change in the regulatory landscape for anatomic pathology. While the rule aims to enhance patient safety and ensure the reliability of diagnostic tests, it also introduces new challenges and costs for clinical laboratories. Understanding and preparing for these changes is essential for maintaining compliance and continuing to provide high-quality patient care. As the medical community navigates this new regulatory environment, ongoing dialogue and collaboration with regulatory bodies will be crucial for addressing emerging issues and ensuring that diagnostic innovation can thrive.
If you have related needs, please feel free to contact us for more information or product support.
Reference
This article is for research use only. Do not use in any diagnostic or therapeutic application.

Cat.No. GP-DQL-00203
Rotavirus Antigen Group A and Adenovirus Antigen Rapid Test Kit (Colloidal Gold)

Cat.No. GP-DQL-00206
Adenovirus Antigen Rapid Test Kit (Colloidal Gold), Card Style

Cat.No. GP-DQL-00207
Adenovirus Antigen Rapid Test Kit (Colloidal Gold), Strip Style

Cat.No. GP-DQL-00211
Rotavirus Antigen Group A Rapid Test Kit (Colloidal Gold), Card Type

Cat.No. GP-DQL-00212
Rotavirus Antigen Group A Rapid Test Kit (Colloidal Gold), Card Type

Cat.No. IP-00189
Influenza A Rapid Assay Kit

Cat.No. GH-DQL-00200
Follicle-stimulating Hormone Rapid Test Kit (Colloidal Gold)
Cat.No. GH-DQL-00201
Insulin-like Growth Factor Binding Protein 1 Rapid Test Kit (Colloidal Gold)

Cat.No. GH-DQL-00202
Luteinizing Hormone Rapid Test Kit (Colloidal Gold)

Cat.No. GH-DQL-00208
Follicle-stimulating Hormone Rapid Test Kit (Colloidal Gold), Strip Style
Cat.No. GH-DQL-00209
Insulin-like Growth Factor Binding Protein 1 Rapid Test Kit(Colloidal Gold), Strip Style

Cat.No. GH-DQL-00210
Luteinizing Hormone Rapid Test Kit (Colloidal Gold), Strip Style
Cat.No. IH-HYW-0001
hCG Pregnancy Test Strip
Cat.No. IH-HYW-0002
hCG Pregnancy Test Cassette
Cat.No. IH-HYW-0003
hCG Pregnancy Test Midstream

Cat.No. GD-QCY-0001
Cocaine (COC) Rapid Test Kit

Cat.No. GD-QCY-0002
Marijuana (THC) Rapid Test Kit
Cat.No. GD-QCY-0003
Morphine (MOP300) Rapid Test Kit
Cat.No. GD-QCY-0004
Methamphetamine (MET) Rapid Test Kit
Cat.No. GD-QCY-0005
Methylenedioxymethamphetamine ecstasy (MDMA) Rapid Test Kit
Cat.No. GD-QCY-0006
Amphetamine (AMP) Rapid Test Kit
Cat.No. GD-QCY-0007
Barbiturates (BAR) Rapid Test Kit

Cat.No. GD-QCY-0008
Benzodiazepines (BZO) Rapid Test Kit
Cat.No. GD-QCY-0009
Methadone (MTD) Rapid Test Kit
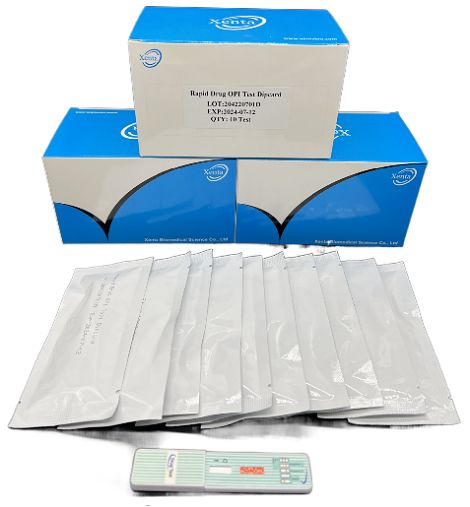
Cat.No. GD-QCY-0011
Opiate (OPI) Rapid Test Kit
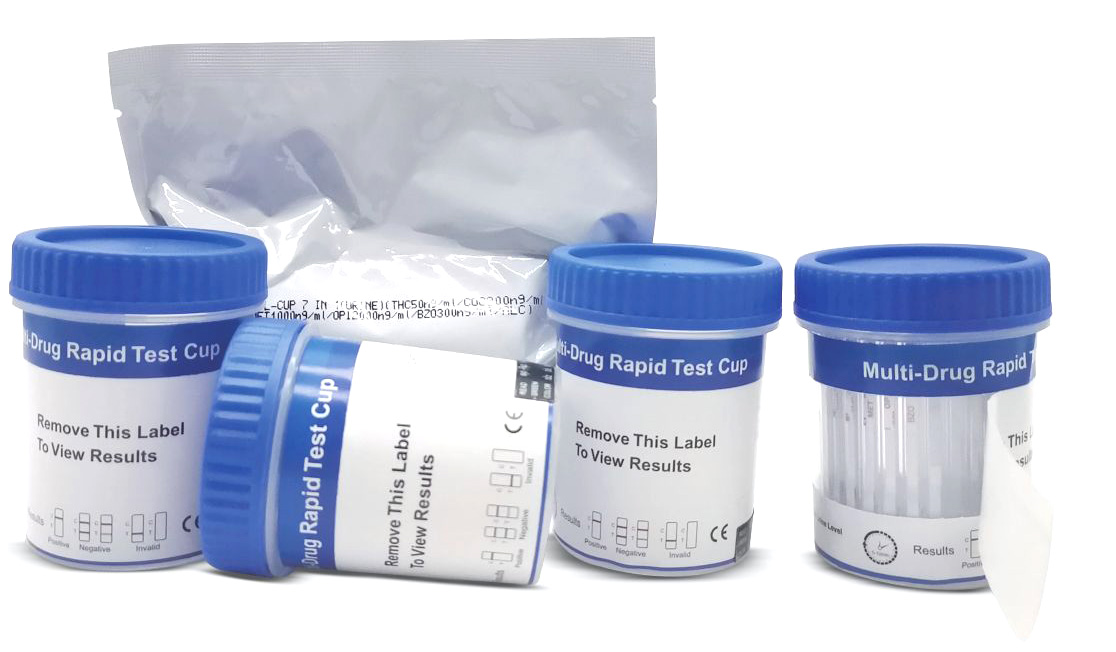
Cat.No. ID-HYW-0002
Multi-Drug Test L-Cup, (5-16 Para)
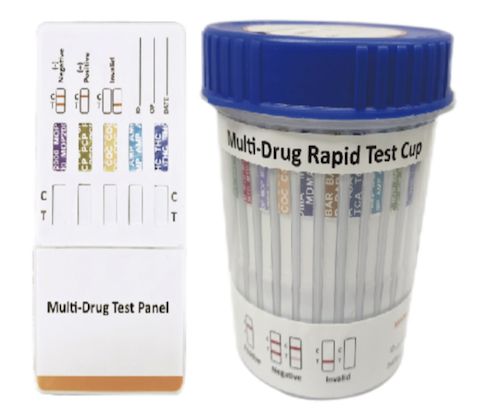
Cat.No. ID-HYW-0005
Multi-Drug Rapid Test (Dipcard & Cup) with Fentanyl
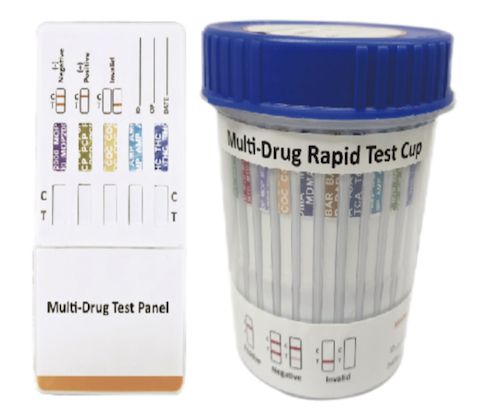
Cat.No. ID-HYW-0006
Multi-Drug Rapid Test (Dipcard & Cup) without Fentanyl
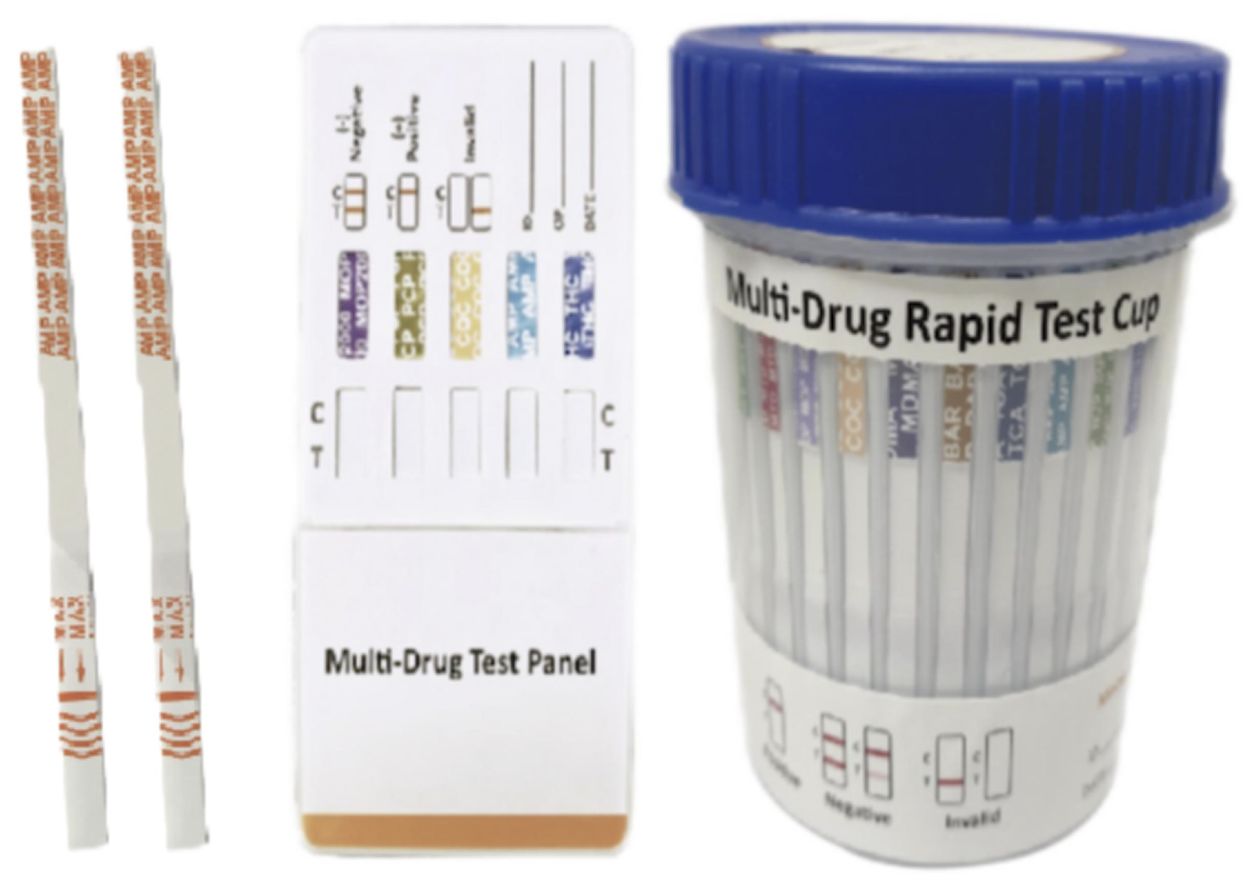
Cat.No. ID-HYW-0007
Multi-Drug 2~14 Drugs Rapid Test (Dipstick & Dipcard & Cup)
Cat.No. ID-HYW-0008
Fentanyl (FYL) Rapid Test (For Prescription Use)
Cat.No. ID-HYW-0009
Fentanyl Urine Test Cassette (CLIA Waived)
Cat.No. ID-HYW-0010
Fentanyl Urine Test Cassette (Home Use)
|
There is no product in your cart. |
Copyright © 2026 Alta DiagnoTech. All rights reserved.