- Home
- Resource
- Disease Diagnosis
- Genetic Diseases
- Standardizing Rett Syndrome (RTT) Diagnosis: A Step-by-Step Genetic and Clinical Pathway
- Home
- IVD
- By Technology Types
- By Diseases Types
- By Product Types
- Research
- Resource
- Distributors
- Company

Rett syndrome (RTT) presents complex diagnostic challenges, requiring a systematic approach that integrates clinical evaluation with advanced genetic testing. This resource outlines a standardized diagnostic pathway for RTT, from recognizing early symptoms to implementing genetic testing strategies and post-diagnostic care.
Rett syndrome (RTT) is a rare, severe neurodevelopmental disorder primarily affecting females, characterized by progressive loss of motor and communication skills, repetitive hand movements, and developmental regression, typically emerging between 6–18 months of age. Most cases are linked to mutations in the MECP2 gene, disrupting normal brain function. While classic RTT follows a distinct clinical trajectory, atypical forms present with variable symptoms, often complicating diagnosis. Early genetic testing and multidisciplinary care are critical for managing symptoms, optimizing quality of life, and guiding family planning.
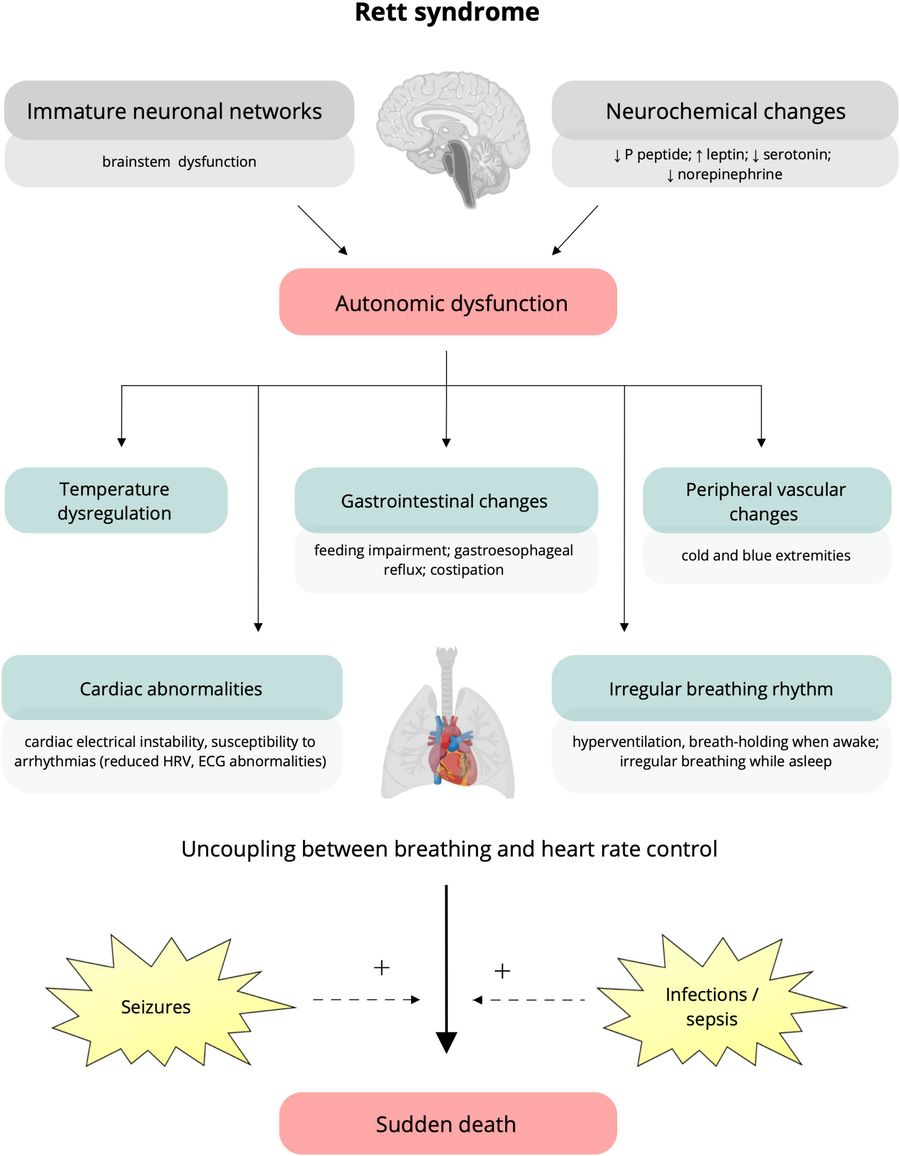 Fig.1 Putative underlying mechanisms and clinical manifestations of autonomic dysfunction in Rett syndrome (RTT). (Cordani R, et al., 2023)
Fig.1 Putative underlying mechanisms and clinical manifestations of autonomic dysfunction in Rett syndrome (RTT). (Cordani R, et al., 2023)
Rett syndrome (RTT) presents with a distinct clinical progression that evolves through four characteristic stages:
Rett syndrome (RTT) should be suspected when an infant (typically female) exhibits developmental regression (loss of speech/hand skills) followed by stereotyped hand movements and breathing irregularities. Additional red flags include decelerated head growth, gait abnormalities, and transient autistic-like features preceding regression. While classic RTT shows this clear trajectory, atypical cases may present with early-onset seizures or preserved speech but still require genetic testing.
A targeted, stepwise genetic testing approach optimizes diagnostic accuracy for Rett syndrome (RTT), beginning with MECP2 analysis followed by expanded testing when needed.
Step 1: Initial MECP2 Testing
First-line testing focuses on the MECP2 gene (identifying ~95% of classic RTT cases) using Sanger sequencing for point mutations and MLPA for deletions/duplications. This cost-effective approach confirms diagnosis in most patients presenting with core features like regression and hand stereotypies.
Step 2: Expanded Gene Panel
For atypical presentations or MECP2-negative cases, testing expands to CDKL5 (early seizure variants) and FOXG1 (congenital-onset), with whole exome sequencing reserved for unresolved cases. This tiered strategy detects ~50% of remaining diagnoses while optimizing resource utilization.
A definitive Rett syndrome (RTT) diagnosis requires correlating clinical features with genetic findings through multidisciplinary evaluation. Neurologists and geneticists should jointly assess the patient's developmental trajectory alongside genetic test results, as ~5-10% of MECP2-positive cases show atypical presentations, while some clinically classic RTT cases may initially test negative due to mosaicism or novel variants. Discordant cases benefit from re-evaluation of both phenotype (e.g., seizure types, growth charts) and genotype (e.g., re-analysis of VUS, additional testing for CDKL5/FOXG1), ensuring comprehensive diagnosis. This integration is particularly crucial for males and atypical cases where early intervention hinges on accurate classification.
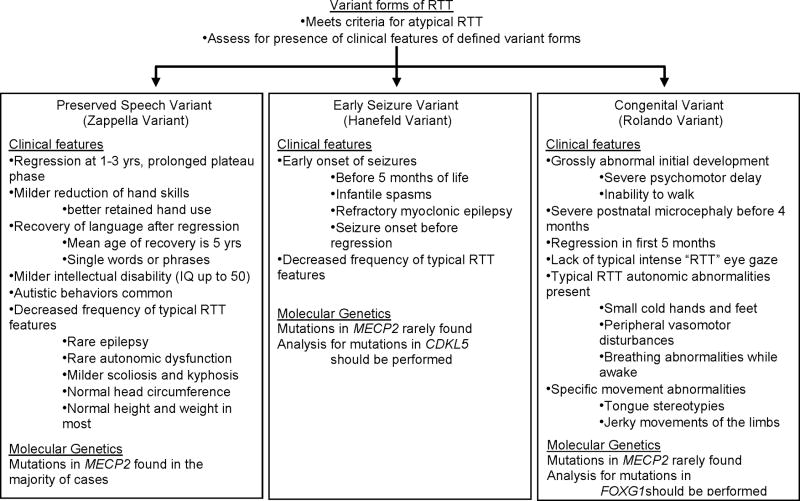 Fig.2 Specific variant forms of Rett syndrome (RTT) flow diagram. (Neul J L, et al., 2010)
Fig.2 Specific variant forms of Rett syndrome (RTT) flow diagram. (Neul J L, et al., 2010)
Following a confirmed Rett syndrome (RTT) diagnosis, a multidisciplinary care plan should be implemented, focusing on symptom management (seizure control, motor dysfunction, gastrointestinal issues), developmental support (communication aids, physical/occupational therapy), and family-centered care (genetic counseling for recurrence risk and psychosocial support). Regular neurological and orthopedic monitoring is essential to address complications like scoliosis or cardiac abnormalities, while emerging therapies targeting MECP2 dysfunction offer hope for future interventions.
Alta DiagnoTech offers advanced genetic testing kits for Rett syndrome (RTT), featuring comprehensive MECP2/CDKL5/FOXG1 analysis with high accuracy, rapid turnaround time, and user-friendly protocols to streamline diagnosis. If you have related needs, please feel free to contact us for more information or product support.
References
This article is for research use only. Do not use in any diagnostic or therapeutic application.
|
There is no product in your cart. |
Copyright © 2026 Alta DiagnoTech. All rights reserved.