- Home
- Resource
- Disease Diagnosis
- Metabolic Diseases
- Preventing Neurocrisis: A Comprehensive Guide to GA1 Diagnosis and Screening
- Home
- IVD
- By Technology Types
- By Diseases Types
- By Product Types
- Research
- Resource
- Distributors
- Company

Glutaric acidemia type I (GA1) is a rare but devastating inherited metabolic disorder where timely diagnosis is the single most critical factor in preventing irreversible neurological damage. This resource provides a comprehensive overview of the diagnostic pathway for GA1, from initial newborn screening to molecular confirmation, highlighting key biomarkers, technologies, and common diagnostic challenges.
Glutaric acidemia type I (GA1) is an inherited metabolic disorder caused by a deficiency of the enzyme glutaryl-CoA dehydrogenase (GCDH), leading to the toxic accumulation of glutaric acid, 3-hydroxyglutaric acid, and glutaryl-CoA in the body. This biochemical imbalance primarily affects the central nervous system, particularly the basal ganglia. While newborns often appear asymptomatic, they are at high risk of experiencing an acute encephalopathic crisis—typically triggered by fever or infection during early childhood—which can result in irreversible striatal damage and severe dystonic movement disorders.
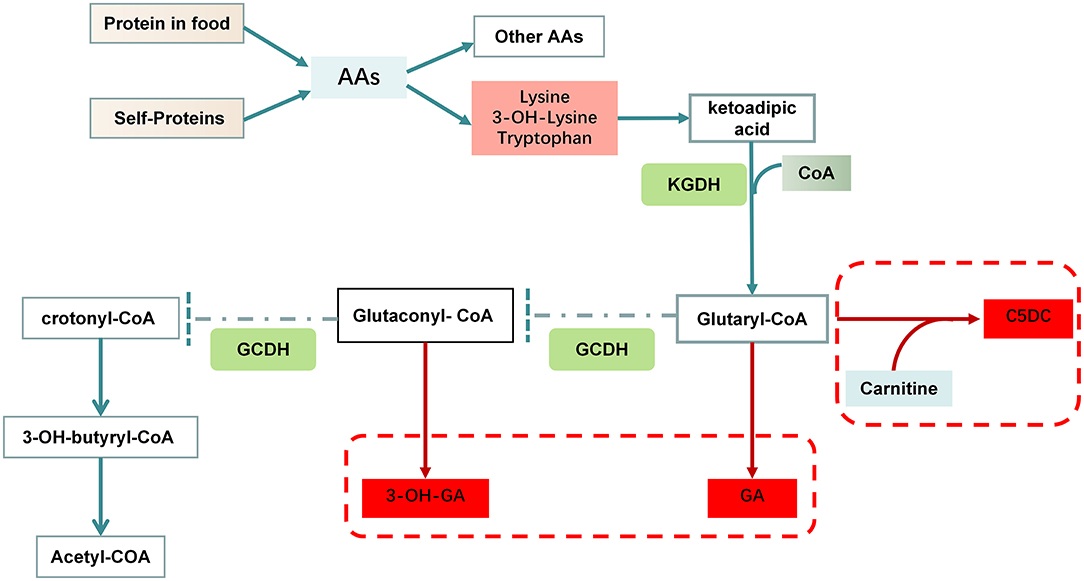 Fig.1 Disorders of lysine and tryptophan metabolism in glutaric acidemia type I (GA1). (Li Q, et al., 2021)
Fig.1 Disorders of lysine and tryptophan metabolism in glutaric acidemia type I (GA1). (Li Q, et al., 2021)
Timely diagnosis of glutaric acidemia type I (GA1) is critical to prevent irreversible neurological damage through pre-symptomatic intervention. The diagnosis of GA1 relies on the detection of specific biochemical biomarkers that accumulate due to glutaryl-CoA dehydrogenase (GCDH) deficiency. These biomarkers can be measured in various biological samples and form the basis of newborn screening, confirmatory testing, and surveillance.
The diagnostic pathway for glutaric acidemia type I (GA1) is a multi-stage process designed to achieve pre-symptomatic detection, which is critical for preventing irreversible neurological damage. This structured approach begins with population-wide screening, progresses through specialized biochemical testing to confirm the diagnosis, and culminates in genetic analysis for definitive characterization and family planning. This systematic workflow ensures high accuracy and enables the prompt initiation of life-saving dietary and medical management.

Newborn Screening (NBS)
Newborn screening (NBS) serves as the critical first tier in identifying infants at risk for GA1. This involves analyzing dried blood spots collected from a heel prick using tandem mass spectrometry (MS/MS) to detect elevated levels of glutarylcarnitine (C5DC). An abnormal NBS result is not diagnostic but acts as an essential trigger for immediate follow-up specialized testing to confirm or rule out the disorder.

Biochemical Confirmation
Biochemical confirmation is the essential second step, typically involving urinary organic acid analysis by gas chromatography-mass spectrometry (GC-MS). This test identifies the pathognomonic pattern of elevated glutaric acid, 3-hydroxyglutaric acid (the most specific marker), and often glutaconic acid. This profile provides definitive biochemical evidence of the enzyme deficiency and is crucial for differentiating GA1 from other metabolic conditions with similar presentations.
Definitive Molecular Diagnosis
Definitive molecular diagnosis is achieved through DNA sequencing of the GCDH gene to identify pathogenic variants. This step confirms the diagnosis at the genetic level, allows for precise carrier testing within the family, enables prenatal diagnosis in future pregnancies, and can sometimes provide prognostic information based on the identified genotype, thus completing the diagnostic journey.
Diagnosing GA1 presents significant challenges due to variable biomarker expression, particularly in "low excretors" who may show normal or borderline levels of key indicators like glutarylcarnitine (C5DC) and urinary organic acids during metabolic stability. Overreliance on newborn screening can be misleading, as normal results do not definitively rule out GA1, and biomarker profiles may overlap with other disorders such as MADD. Additionally, interpreting genetic variants of uncertain significance (VUS) in the GCDH gene requires careful analysis. A definitive diagnosis therefore necessitates integrating biochemical testing during symptomatic periods, clinical evaluation, and genetic sequencing to avoid misdiagnosis and ensure accurate detection.
The future of glutaric acidemia type I (GA1) diagnostics is moving towards integrated, multi-omics platforms that combine second-tier genetic sequencing with newborn screening to reduce false positives and identify variants of uncertain significance (VUS) more efficiently. Advanced technologies like next-generation sequencing (NGS) and machine learning-based biomarker analysis will enable earlier and more precise detection, even in low-excretor cases, while emerging techniques such as liquid chromatography-high-resolution mass spectrometry (LC-HRMS) could allow simultaneous quantification of multiple metabolites and biomarkers. These advancements will ultimately support personalized treatment strategies and improve long-term outcomes for GA1 patients.
Alta DiagnoTech provides comprehensive IVD solutions for glutaric acidemia type I (GA1), spanning newborn screening kits, confirmatory biochemical assays, and definitive genetic tests. If you have related needs, please feel free to contact us for more information or product support.
Reference
This article is for research use only. Do not use in any diagnostic or therapeutic application.
|
There is no product in your cart. |
Copyright © 2026 Alta DiagnoTech. All rights reserved.