Niemann-Pick disease (NPD) is a rare, autosomal recessive lysosomal storage disorder. This resource provides a comprehensive overview of the modern diagnostic landscape for NPD, which has evolved beyond traditional sequential testing to an era of precision diagnostics, where a definitive diagnosis is achieved through the robust integration of three core pillars: enzyme activity assays to confirm functional defects, advanced biomarker analysis to provide rapid screening and monitoring capabilities, and genetic analysis to provide definitive molecular confirmation.
Introduction to Niemann-Pick Disease (NPD)
Niemann-Pick disease (NPD) is a group of rare, inherited lysosomal storage disorders characterized by the harmful accumulation of lipids, particularly sphingomyelin and cholesterol, within cells throughout the body. This buildup results from specific genetic defects that disrupt essential metabolic pathways, leading to progressive cellular dysfunction and damage to major organs such as the liver, spleen, lungs, and, in many forms, the central nervous system. The disease encompasses several types—primarily A, B, and C—each with distinct genetic causes, ages of onset, and clinical severity, ranging from a severe infantile form with profound neurodegeneration to later-onset forms with primarily systemic manifestations.
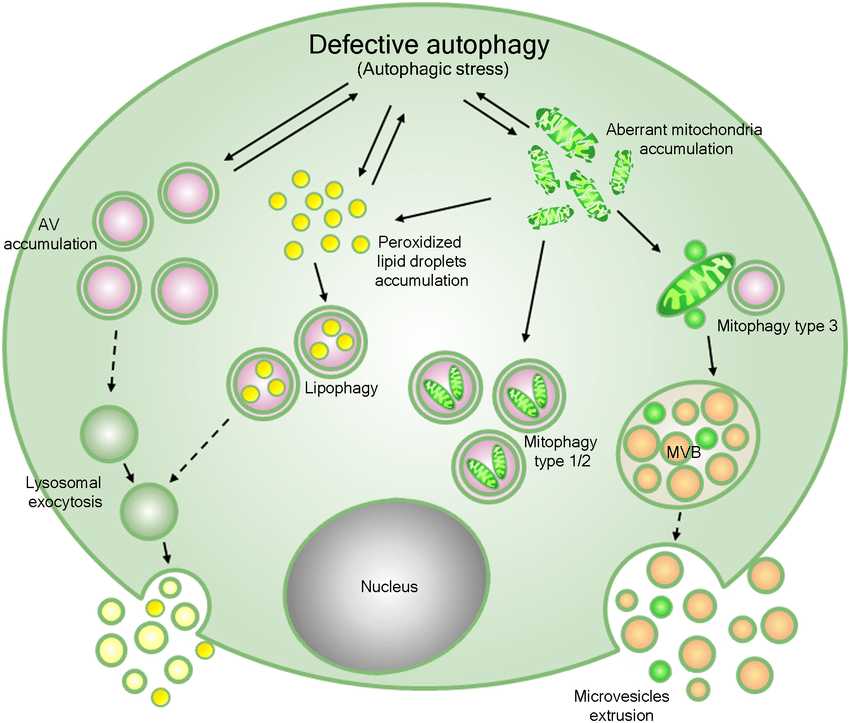 Fig.1 A proposed model for the pathogenesis of Niemann-Pick disease (NPD) type B. (Canonico, et al., 2016)
Fig.1 A proposed model for the pathogenesis of Niemann-Pick disease (NPD) type B. (Canonico, et al., 2016)
Enzyme Activity Assay for Niemann-Pick Disease (NPD)
The definitive biochemical diagnosis for Niemann-Pick disease (NPD) hinges on the enzyme activity assay, a test that directly measures the functional capacity of specific lysosomal enzymes in patient cells, most commonly from a blood sample. This assay serves as the gold standard for confirming NPD types A and B by identifying a profound deficiency in acid sphingomyelinase (ASM) activity, which is the fundamental enzymatic defect that disrupts sphingomyelin metabolism and leads to pathological lipid accumulation.
Diagnostic Biomarkers for Niemann-Pick Disease (NPD)
Diagnostic biomarkers are measurable molecules that provide a reliable indicator of a disease's presence or activity. For Niemann-Pick disease (NPD), the discovery and validation of specific biomarkers have revolutionized the diagnostic workflow, offering powerful tools for screening, supporting diagnosis, and monitoring disease progression. The most significant advances have been made with biomarkers for NPD Types A and B:
Lyso-Sphingomyelin (Lyso-SM)
This deacylated form of sphingomyelin accumulates dramatically in the plasma of individuals with ASM deficiency. Its elevated levels serve as a highly sensitive and specific biochemical signature for NPD Types A and B.
Lyso-Sphingomyelin-509 (Lyso-SM-509)
An oxidized derivative of Lyso-SM, this biomarker is also markedly elevated in NPD patients. The ratio of Lyso-SM-509 to Lyso-SM can provide additional diagnostic information and is particularly useful for differentiating NPD from other conditions.
Genetic Analysis of Niemann-Pick Disease (NPD)
Genetic analysis is the definitive method for confirming the molecular diagnosis of Niemann-Pick disease (NPD) and is essential for understanding its specific subtype, inheritance pattern, and implications for family members. This process involves identifying pathogenic mutations in the genes known to cause NPD, which provides the most precise explanation for the biochemical and clinical findings. The specific genes analyzed depend on the suspected subtype:
SMPD1 Gene Analysis (for Types A & B)
Sequencing the SMPD1 gene is performed to identify disease-causing mutations in individuals with confirmed or suspected acid sphingomyelinase (ASM) deficiency. Identifying biallelic pathogenic variants in this gene provides a conclusive diagnosis for NPD Types A and B and enables accurate carrier testing within the family.
NPC1 and NPC2 Gene Analysis (for Type C)
For patients suspected of having the neurologically progressive NPD Type C, genetic testing focuses on the NPC1 and NPC2 genes. The vast majority (over 95%) of Type C cases are caused by mutations in the NPC1 gene, with a small fraction linked to NPC2. Identifying biallelic pathogenic variants in either gene confirms the diagnosis.
IVD Products for Niemann-Pick Disease (NPD)
Alta DiagnoTech is dedicated to advancing the diagnosis of Niemann-Pick disease (NPD) by providing a comprehensive portfolio of in vitro diagnostic (IVD) solutions. Our integrated product line is designed to support clinical laboratories at every stage of the diagnostic journey, from initial high-throughput screening to definitive confirmation, enabling a precise and efficient path to diagnosis for patients. If you have related needs, please feel free to contact us for more information or product support.
| Product Category |
Product Name |
Application |
| Newborn Screening |
Lyso-SM Screening Assay Kit (DBS) |
First-tier newborn screening and rapid patient triage using dried blood spots (DBS). |
| Enzymatic Confirmation |
Acid Sphingomyelinase (ASM) Activity Detection Kit |
Definitive confirmation of NPD Types A and B. |
| Biomarker Profiling |
Lyso-SM/Lyso-SM-509 Quantitative Detection Kit (LC-MS/MS) |
Specific biomarker profiling to support diagnosis, aid in subtyping, and monitor disease progression. |
| Genetic Confirmation |
Niemann-Pick disease (NPD) Genetic Testing Panel |
Molecular genetic confirmation and carrier testing for NPD Types A, B, and C. |
Reference
- Canonico, Barbara, et al. "Defective autophagy, mitochondrial clearance and lipophagy in niemann-pick type B lymphocytes." PloS one 11.10 (2016): e0165780.
This article is for research use only. Do not use in any diagnostic or therapeutic application.