Companion diagnostics (CDx) have become indispensable tools in precision medicine, serving as the bridge between targeted therapies and the patients who stand to benefit most. Defined as devices essential for the safe and effective use of corresponding medicinal products, CDx identify individuals with specific genetic or molecular profiles likely to respond positively to a given treatment. Examples range from Abbott's RealTime IDH1 assay, which selects acute myeloid leukemia patients for Tibsovo therapy, to Agilent's PD-L1 IHC 22C3 pharmDx, used to determine eligibility for Keytruda in non-small cell lung cancer.
While pre-marketing clinical trials establish initial safety and efficacy, post-marketing validation ensures these devices maintain performance over time in real-world settings. This ongoing assessment addresses gaps in pre-approval data, such as long-term stability, performance across diverse populations, and detection of rare adverse events. For instance, complex CDx development processes can lead to variability in results post-launch, making continuous monitoring essential to uphold the quality of personalized care. Post-marketing data also informs risk management strategies, updates to product labeling, and adjustments to clinical guidelines, directly impacting patient outcomes.
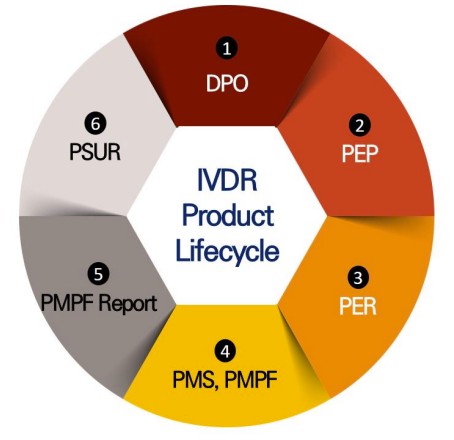 Fig.1 IVDR safety and performance verification process. (Kang S. L., et al., 2024)
Fig.1 IVDR safety and performance verification process. (Kang S. L., et al., 2024)
Regulatory Frameworks: A Global Overview
Regulatory bodies worldwide have implemented distinct systems to oversee post-marketing validation of CDx, reflecting varying priorities and approaches to risk management. The U.S. Food and Drug Administration (FDA), European Medicines Agency (EMA), Japan's Pharmaceuticals and Medical Devices Agency (PMDA), and South Korea's Ministry of Food and Drug Safety (MFDS) each enforce unique requirements, though all aim to ensure long-term device reliability.
A key distinction lies in scope: the EMA mandates post-marketing surveillance for all CDx, while the FDA, PMDA, and MFDS target devices deemed high-risk or with insufficient pre-marketing data. Additionally, reporting mechanisms, data requirements, and timelines differ, creating a complex landscape for manufacturers operating internationally.
United States: FDA's Post-Approval Studies (PAS)
The FDA's post-marketing framework centers on Post-Approval Studies (PAS), authorized under Title 21 of the Code of Federal Regulations. PAS are required when pre-marketing data is incomplete—for example, when uncertainties exist about a CDx's performance in patients with rare biomarkers or long-term use. These studies collect specific data to address lingering questions about safety and effectiveness, with a focus on real-world performance rather than controlled trial settings.
Key Requirements for PAS:
Study Design: Must include clear objectives, statistically sound sample sizes, and defined endpoints (e.g., false positive rates, patient response correlations).
Reporting: Manufacturers submit an interim report to update progress and a final report summarizing results, compliance with protocols, and any deviations.
Focus Areas: For CDx, PAS often includes data on biomarker-negative patients, a subgroup frequently underrepresented in pre-marketing trials.
A notable example is the MK-3475 MSI-H FMI F1CDx Post Approval Analysis, which tracked 41 patients with advanced microsatellite instability-high (MSI-H) solid tumors. The study confirmed a 42.9% objective response rate to pembrolizumab in CDx-positive patients, aligning with pre-approval findings and reinforcing the device's clinical utility.
European Union: EMA's Integrated Post-Market Surveillance (PMS)
Under the In Vitro Diagnostic Regulation (IVDR), the EMA requires all CDx (classified as high-risk Class C devices) to implement Post-Market Surveillance (PMS) as part of their quality management systems. PMS integrates ongoing data collection, analysis, and reporting to monitor device performance throughout its lifecycle.
Core Components of EMA’s System:
PMS Plan: Documents strategies for collecting safety and performance data from clinical use, user feedback, and literature reviews.
Post-Market Performance Follow-Up (PMPF): A mandatory subset of PMS for Class C devices, focusing on verifying long-term accuracy and detecting emerging risks. PMPF plans must include analyses to ensure the benefit-to-risk ratio remains favorable.
Periodic Safety Update Reports (PSUR): Annual submissions summarizing key findings, device sales data, and population characteristics (e.g., usage frequency across regions). PSURs also include assessments of new risks and corrective actions taken.
A challenge in the EU framework is the delayed rollout of EUDAMED, the centralized database for medical device information, which limits transparency and public access to post-marketing data. Despite this, the EMA's holistic approach ensures continuous monitoring of all CDx, minimizing gaps in oversight.
Japan: PMDA's PMS and Re-Evaluation
Japan's post-marketing system combines Post-Market Surveillance (PMS) with targeted Re-Evaluation to ensure CDx reliability. PMS is mandatory for all manufacturers, requiring the collection of real-world data on quality, safety, and effectiveness through three methods: use history investigations, post-marketing database research, and clinical trials.
Key Features:
PMS Activities: Manufacturers must maintain detailed operating procedures for data collection, including patient demographics, test results, and adverse events.
Re-Evaluation: Conducted for devices designated by the Ministry of Health, Labor and Welfare (MHLW), this process assesses long-term performance using real-world evidence, such as patient drug response data.
An illustrative case is the PMS for F1CDx in Japan, which evaluated its utility in identifying patients with recurrent adenoid cystic carcinoma for immunotherapy. The study included a 5-year follow-up period to track objective response rates and disease control, ensuring alignment with pre-marketing claims.
South Korea: MFDS's Re-Evaluation and Renewal
The MFDS employs two complementary systems: Re-Evaluation and Renewal. Re-Evaluation targets CDx requiring reassessment due to safety concerns or advances in scientific knowledge, while Renewal mandates periodic reviews every 5 years to ensure compliance with the latest standards.
Requirements for Re-Evaluation:
Data Submission: Includes adverse event analyses, clinical trial updates, and literature reviews (e.g., new findings on biomarker accuracy).
Safety Information: Must cover four categories: domestic and foreign academic papers, clinical trial data, foreign manufacturer manuals, and government agency announcements.
Renewal Process:
Manufacturers submit data on storage stability, analytical performance, and quality control 180 days before expiration of the device's approval. A successful review extends the validity period, ensuring ongoing alignment with scientific advancements.
A practical example is the re-evaluation of a CDx for non-small cell lung cancer, which incorporated new data from international trials showing reduced accuracy in smokers, leading to updated usage guidelines and labeling.
Comparative Analysis: Key Differences and Commonalities
| Regulator |
System |
Scope |
Data Focus |
Reporting Timelines |
| FDA |
Post-Approval Studies |
High-risk devices with gaps in pre-marketing data |
Real-world performance, biomarker-negative patients |
Interim and final reports as specified |
| EMA |
PMS + PMPF + PSUR |
All Class C CDx |
Benefit-risk ratio, emerging risks |
Annual PMPF and PSUR reports |
| PMDA |
PMS + Re-Evaluation |
All devices, targeted re-evaluation for high-priority CDx |
Real-world clinical outcomes |
Ongoing PMS; re-evaluation as designated |
| MFDS |
Re-Evaluation + Renewal |
High-risk devices; 5-year reviews |
Compliance with the latest scientific standards |
180-day pre-expiration renewal submissions |
Common goals include detecting long-term risks, verifying effectiveness, and supporting risk management, while differences reflect regional priorities: the EMA emphasizes universal oversight, the FDA focuses on targeted data gaps, and Asian regulators prioritize alignment with evolving scientific standards.
Future Directions: Harmonization and Innovation
As CDx technology advances, global regulators are working toward greater harmonization. The International Medical Device Regulators Forum (IMDRF) has developed guidelines to streamline post-marketing requirements, reducing burdens for manufacturers and ensuring consistent patient protection. Key areas for progress include:
- Data Transparency: Expanding access to databases like EUDAMED to facilitate cross-border collaboration.
- Real-World Evidence Integration: Leveraging large-scale clinical datasets to enhance post-marketing validation efficiency.
- Adaptability: Updating regulations to keep pace with next-generation CDx, such as multi-gene panels and liquid biopsies.
These efforts will be critical in ensuring that CDx continues to deliver on the promise of precision medicine, providing safe, effective, and personalized care to patients worldwide.
In summary, post-marketing validation is a cornerstone of CDx regulation, with global systems tailored to regional needs yet united in their commitment to patient safety. By understanding these frameworks, manufacturers can navigate compliance challenges, while clinicians and patients can trust in the reliability of these life-saving diagnostic tools.
If you have related needs, please feel free to contact us for more information or product support.
Reference
- Kang, Su Lim, Ji Yean Kwon, and Sung Min Kim. "Insights into post-marketing clinical validation of companion diagnostics with reference to the FDA, EMA, PMDA, and MFDS." Molecular Therapy Methods & Clinical Development 32.4 (2024).
This article is for research use only. Do not use in any diagnostic or therapeutic application.
Trending Products