Mucopolysaccharidoses (MPS) are a group of inherited metabolic disorders caused by deficient lysosomal enzymes leading to glycosaminoglycan (GAG) accumulation. This resource provides a comprehensive overview of the diagnostic pathway for MPS, detailing the essential biomarkers, enzymatic assays, and molecular genetic analyses that enable accurate diagnosis and subtyping. We will explore the specific IVD products and methodologies that support this multi-tiered diagnostic approach, providing clinicians and laboratories with the tools needed for effective patient management.
Introduction to Mucopolysaccharidoses (MPS)
Mucopolysaccharidoses (MPS) are a group of rare, inherited lysosomal storage disorders caused by deficiencies in the enzymes needed to break down complex carbohydrates called glycosaminoglycans (GAGs). This enzymatic defect leads to the progressive accumulation of GAGs within cells, resulting in widespread tissue and organ damage. The spectrum of MPS encompasses multiple subtypes, each with a distinct genetic cause, but collectively they often present with characteristic skeletal abnormalities, coarse facial features, and involvement of the cardiovascular, respiratory, and central nervous systems.
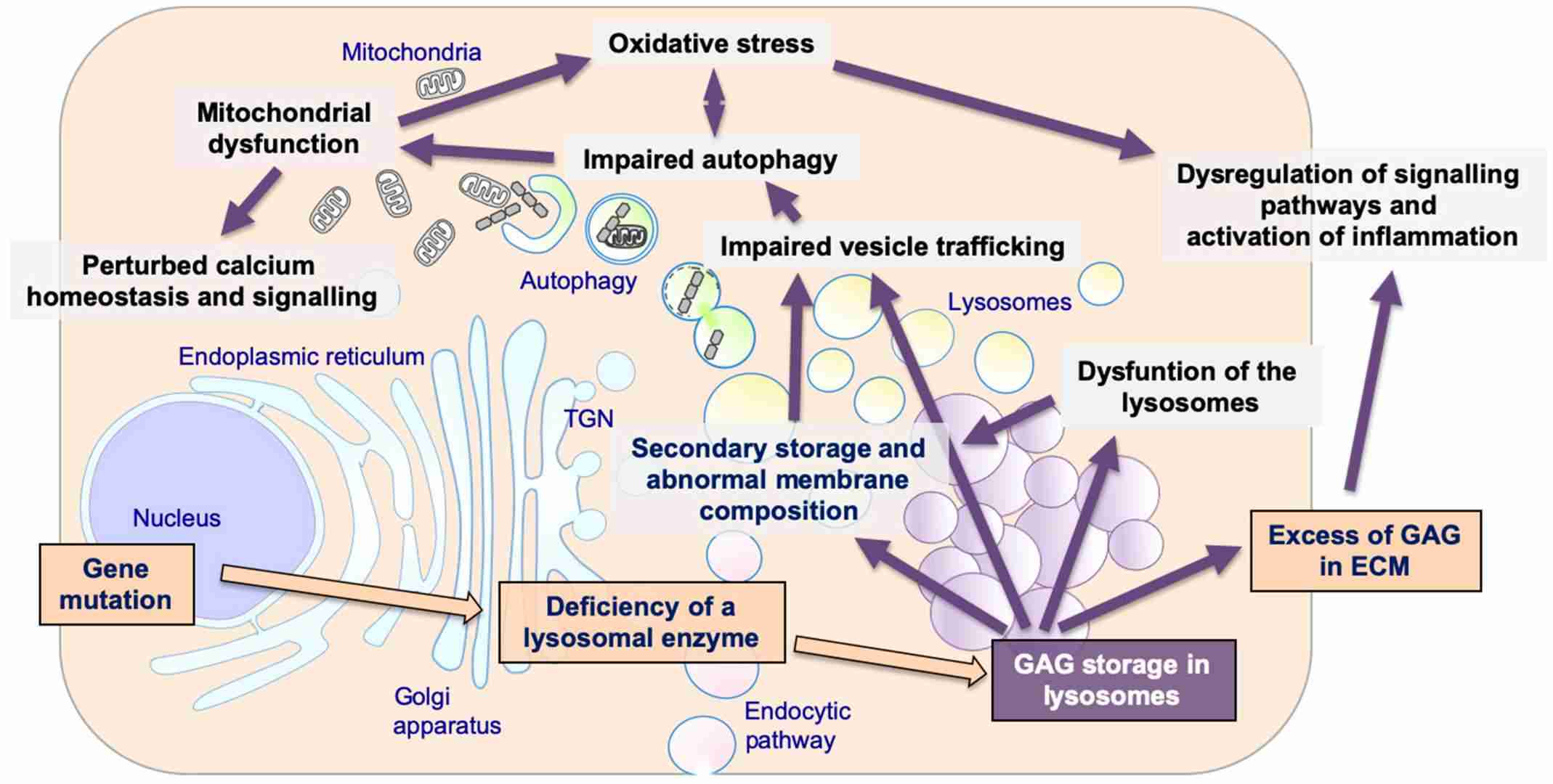 Fig.1 The pathogenetic cascade of mucopolysaccharidoses (MPSs). (Fecarotta S, et al., 2020)
Fig.1 The pathogenetic cascade of mucopolysaccharidoses (MPSs). (Fecarotta S, et al., 2020)
Diagnostic Biomarkers for Mucopolysaccharidoses (MPS)
The laboratory diagnosis of mucopolysaccharidoses (MPS) critically relies on the detection and analysis of specific biomarkers, primarily the glycosaminoglycans (GAGs) that accumulate due to enzymatic deficiencies. The profile of elevated GAGs in a patient's urine or blood provides critical clues about the underlying enzymatic defect, effectively guiding subsequent confirmatory enzyme activity and genetic testing for a precise diagnosis.
Dermatan Sulfate (DS)
Dermatan sulfate (DS) accumulates due to deficiencies in enzymes such as iduronidase (MPS I) and iduronate-2-sulfatase (MPS II). Its buildup is clinically associated with profound somatic manifestations, including coarse facial features, joint contractures, skeletal dysplasia (dysostosis multiplex), and organomegaly. The detection of elevated DS, particularly when combined with heparan sulfate, is a classic biomarker signature pointing towards MPS I, II, or VII.
Heparan Sulfate (HS)
Heparan sulfate (HS) accumulation is a hallmark of enzyme deficiencies affecting its degradation pathway, most notably in MPS III (Sanfilippo syndrome). Its primary clinical significance is its strong association with severe neurological degeneration. Elevated HS in the central nervous system leads to progressive intellectual disability, developmental regression, and behavioral disturbances. Isolated HS elevation strongly suggests MPS III, while its co-elevation with DS points to MPS I, II, or VII.
Keratan Sulfate (KS)
Keratan sulfate (KS) is the primary biomarker for MPS IV, also known as Morquio syndrome (types A and B). Its accumulation is uniquely linked to a specific skeletal phenotype characterized by severe skeletal dysplasia, short stature, and ligamentous laxity, without the neurological involvement seen in other types. The detection of significantly elevated KS is therefore highly pathognomonic for MPS IV and directs diagnostic focus towards the GALNS or GLB1 genes.
Chondroitin Sulfate (CS)
Chondroitin sulfate (CS) is a secondary biomarker that can be elevated in several MPS types, often alongside DS. While its isolated, significant elevation is less common, specific patterns of CS chains can provide supportive diagnostic information. Its presence is frequently noted in MPS I, II, and VI, and its analysis can contribute to a more comprehensive biochemical profiling of the disease.
Enzyme Activity Assays for Mucopolysaccharidoses (MPS)
Enzyme activity assays serve as the definitive biochemical method for diagnosing mucopolysaccharidoses (MPS) by directly measuring the functional deficiency of specific lysosomal enzymes responsible for glycosaminoglycan (GAG) degradation. These assays provide conclusive evidence of the underlying enzymatic defect, enabling accurate subtyping of MPS disorders and guiding targeted treatment strategies. Two highly reliable and widely used methodologies for these assays are fluorometric assays and tandem mass spectrometry (MS/MS) assays.
Fluorometric Assays
Fluorometric assays utilize synthetic substrates labeled with a fluorescent group. When the target enzyme cleaves its specific substrate, the fluorescent molecule is released, producing a measurable signal directly proportional to enzyme activity. This method is highly sensitive, cost-effective, and suitable for high-throughput screening, making it a cornerstone in initial MPS diagnostic workflows.
Tandem Mass Spectrometry (MS/MS) Assays
Tandem mass spectrometry (MS/MS) assays employ substrates designed to release specific molecular products detectable by mass spectrometry. This method offers exceptional specificity and precision, allowing simultaneous multiplex analysis of multiple enzyme activities from a single sample. Its ability to minimize interference and provide quantitative results makes it invaluable for confirming diagnoses and comprehensively profiling enzymatic deficiencies.
Molecular Genetic Analyses for Mucopolysaccharidoses (MPS)
Molecular genetic analysis represents the definitive level of diagnosis in mucopolysaccharidoses (MPS), enabling precise identification of the underlying pathogenic variants in specific genes responsible for impaired glycosaminoglycan (GAG) metabolism. This approach not only confirms the diagnosis established through biochemical testing but also enables accurate subtyping, carrier detection, prenatal diagnosis, and informed genetic counseling for affected families. The following genetic testing methodologies are central to this process.
Targeted Gene Sequencing
This methodology focuses on analyzing specific genes known to be associated with MPS, such as IDUA (MPS I) or IDS (MPS II). Using Sanger sequencing or next-generation sequencing (NGS) panels, it efficiently identifies point mutations, small insertions, or deletions within the coding regions of these genes. Targeted sequencing is highly accurate and cost-effective for confirming a diagnosis when biochemical tests strongly suggest a particular MPS subtype.
Next-Generation Sequencing (NGS) Panels
NGS panels allow for the simultaneous sequencing of a curated set of genes related to MPS and other lysosomal storage disorders. This approach is particularly valuable for cases where the clinical presentation or biochemical results are ambiguous, as it can detect variants across multiple genes in a single test. By providing a broad molecular overview, NGS panels facilitate comprehensive differential diagnosis and the identification of rare or unexpected subtypes.
Multiplex Ligation-dependent Probe Amplification (MLPA)
MLPA is a specialized technique used to detect large-scale genomic alterations that are not identifiable by standard sequencing methods. This includes exon-level deletions or duplications within MPS-related genes. MLPA serves as a complementary test to sequencing, ensuring a comprehensive genetic diagnosis by uncovering copy number variations that could explain the disease in cases where sequencing results are inconclusive.
IVD Products for Mucopolysaccharidoses (MPS)
Alta DiagnoTech provides comprehensive IVD solutions for the complete diagnostic workflow of mucopolysaccharidoses (MPS), supporting laboratories from initial screening to definitive genetic confirmation. Our product portfolio includes advanced kits for biomarker analysis, enzymatic activity assays, and molecular genetic testing, enabling accurate and efficient diagnosis of all MPS subtypes. If you have related needs, please feel free to contact us for more information or product support.
| Product Category |
Product Name |
Application & Key Features |
| Biomarker Analysis |
Urinary GAGs Quantitative Assay Kit |
Colorimetric measurement of total glycosaminoglycans for initial screening |
| GAGs Speciation Profiling Kit (LC-MS/MS) |
Simultaneous quantification of DS, HS, KS, and CS for subtype differentiation |
| Enzyme Activity Assays |
MPS Enzyme Fluorometric Panel |
Multiplex assay for IDUA, IDS, GALNS activities using fluorescent substrates |
| MPS Enzyme MS/MS Panel |
Quantitative multiplex assay for MPS I, II, III, IV, VI, and VII enzymes |
| Dried Blood Spot Enzyme Screening Kit |
High-throughput screening for multiple lysosomal enzymes including MPS-related |
| Molecular Genetic Testing |
MPS Targeted NGS Panel |
Comprehensive sequencing of all MPS-related genes (IDUA, IDS, SGSH, etc.) |
| MPS MLPA Analysis Kit |
Detection of exon-level deletions/duplications in major MPS genes |
| MPS Single-Gene Sequencing Kits |
Individual test kits for specific MPS genes (e.g., IDUA, IDS) |
References
- Fecarotta S, Tarallo A, Damiano C, et al. Pathogenesis of mucopolysaccharidoses, an update[J]. International Journal of Molecular Sciences, 2020, 21(7): 2515.
- Kubaski F, de Oliveira Poswar F, Michelin-Tirelli K, et al. Diagnosis of mucopolysaccharidoses[J]. Diagnostics, 2020, 10(3): 172.
This article is for research use only. Do not use in any diagnostic or therapeutic application.