Transthyretin amyloid cardiomyopathy (ATTR-CM) is an underdiagnosed and progressive form of heart failure caused by the deposition of misfolded transthyretin protein in the heart muscle. This resource details the modern, non-invasive diagnostic pathway that has revolutionized its identification, moving from initial clinical suspicion to definitive confirmation. We will systematically explore the role of echocardiography and cardiac MRI in raising suspicion, the critical algorithm of biomarkers and scintigraphy for diagnosis, and the importance of genetic testing.
Introduction to Transthyretin Amyloid Cardiomyopathy (ATTR-CM)
Transthyretin amyloid cardiomyopathy (ATTR-CM) is an underdiagnosed and progressive form of infiltrative heart disease caused by the deposition of unstable transthyretin protein as amyloid fibrils in the myocardial tissue. This deposition leads to increased ventricular wall thickness and stiffness, resulting in heart failure with preserved ejection fraction (HFpEF) and significant morbidity and mortality. The condition manifests in two primary forms: wild-type ATTR-CM, an age-related sporadic disease, and hereditary ATTR-CM, caused by mutations in the TTR gene. The diagnosis of ATTR-CM has undergone a revolutionary shift, evolving from reliance on invasive endomyocardial biopsy to contemporary non-invasive algorithms that integrate nuclear imaging, advanced cardiac imaging, and specific biomarker profiling.
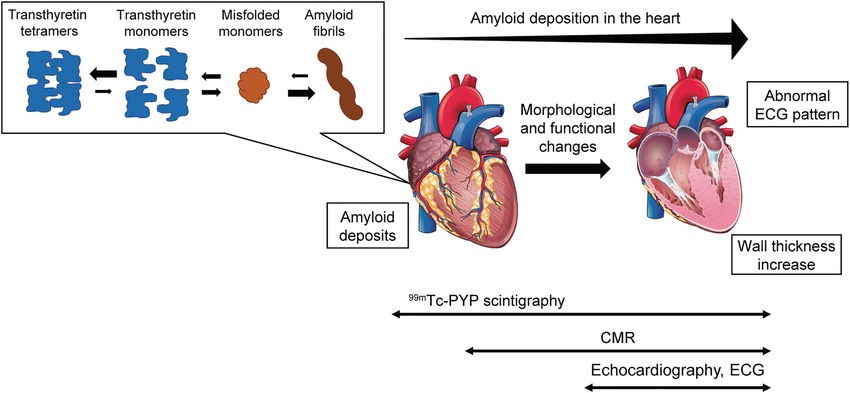 Fig.1 Conceptual diagram for the pathophysiology of transthyretin amyloid cardiomyopathy (ATTR-CM) and applicability of non-invasive diagnostic techniques. (Tahara N, et al., 2022)
Fig.1 Conceptual diagram for the pathophysiology of transthyretin amyloid cardiomyopathy (ATTR-CM) and applicability of non-invasive diagnostic techniques. (Tahara N, et al., 2022)
The Foundation: Raising Suspicion with Imaging & Biomarkers
The initial diagnosis of transthyretin amyloid cardiomyopathy (ATTR-CM) begins with a high index of clinical suspicion, which is then systematically investigated through a combination of characteristic imaging findings and biomarker profiles. This foundational stage is crucial for identifying the cardiac amyloid phenotype and prompting further specific testing, serving as the essential trigger for the non-invasive diagnostic cascade.
Echocardiography
Echocardiography serves as the primary imaging tool for initial assessment, typically revealing increased left ventricular wall thickness with preserved ejection fraction. Characteristic findings include a distinctive "apical-sparing" pattern on global longitudinal strain imaging and a granular sparkling appearance of the myocardium, which together provide strong circumstantial evidence of infiltrative disease.
Cardiac Magnetic Resonance (CMR)
Cardiac magnetic resonance (CMR) provides advanced tissue characterization through late gadolinium enhancement typically demonstrating diffuse patterns with difficulty nulling the blood pool. Quantitative techniques like T1 mapping and extracellular volume fraction measurement directly demonstrate diffuse interstitial expansion, offering high specificity for amyloid infiltration and helping differentiate ATTR-CM from other causes of myocardial thickening.
Core Biomarkers
Core biomarkers play a complementary role in supporting the diagnosis and assessing disease severity. Natriuretic peptides (BNP/NT-proBNP) are consistently elevated due to myocardial wall stress, while high-sensitivity cardiac troponins indicate ongoing subclinical myocyte injury, together providing important prognostic information and helping quantify cardiac involvement.
The Cornerstone of Non-Invasive Diagnosis: The Diagnostic Algorithm
The modern diagnosis of transthyretin amyloid cardiomyopathy (ATTR-CM) is anchored in a validated, stepwise algorithm that has revolutionized clinical practice by enabling definitive diagnosis without routine endomyocardial biopsy. This structured pathway efficiently integrates clinical assessment, laboratory testing, and imaging to accurately identify the disease while critically excluding key mimics. The process is built upon three essential steps that guide the clinician from suspicion to confirmation.
Step 1: Confirm Cardiac Amyloid Phenotype
This initial step involves establishing a pattern of cardiac involvement consistent with an infiltrative process, primarily using echocardiography to detect increased wall thickness with a preserved ejection fraction and a characteristic "apical-sparing" pattern on strain imaging. Cardiac MRI provides further support by identifying a typical pattern of late gadolinium enhancement and an elevated extracellular volume, collectively building a compelling case for an amyloid heart.
Step 2: Rule Out Light-Chain (AL) Amyloidosis
This is a critical and mandatory safety step to exclude the more aggressive AL amyloidosis, which can present with a similar cardiac phenotype. It requires highly sensitive blood tests, specifically the serum free light chain (FLC) assay and immunofixation electrophoresis. A negative result for a monoclonal protein effectively rules out AL amyloidosis and allows the diagnostic process to proceed to the final confirmatory step for ATTR-CM.
Step 3: Confirm ATTR Amyloid with Bone Scintigraphy
The definitive, non-invasive diagnosis is achieved using technetium-99m labeled bone-avid scintigraphy (e.g., PYP, DPD). Intense grade 2 or 3 cardiac uptake on this scan, in the absence of a monoclonal protein, is now internationally recognized as diagnostic for ATTR-CM, eliminating the need for a biopsy in the vast majority of cases and confirming the diagnosis with high specificity.
Completing the Picture: Genotyping and Typing
Once a diagnosis of transthyretin amyloid cardiomyopathy (ATTR-CM) is confirmed non-invasively, the diagnostic process is not yet complete. A final, crucial phase involves precisely characterizing the disease subtype, which carries profound implications for family management, prognosis, and, in some cases, therapy selection. This stage, which completes the patient's diagnostic picture, primarily relies on genetic testing and, in rare specific scenarios, still utilizes biopsy.
- Genetic testing is an essential step for all confirmed ATTR-CM patients to differentiate between the wild-type (wtATTR) and hereditary (hATTR) forms of the disease. Identifying a pathogenic TTR gene mutation confirms hATTR and necessitates genetic counseling and cascade screening of family members. A negative genetic test establishes the diagnosis as wtATTR, which is an acquired, age-related condition.
- Biopsy with histopathological analysis has been largely replaced by the non-invasive algorithm but retains a critical role in resolving diagnostic ambiguity. Its primary use is in cases where the diagnostic pathway is inconclusive, most notably when there is a concomitant positive bone scintigraphy scan and a monoclonal protein in the blood, requiring tissue confirmation to distinguish between ATTR-CM and AL amyloidosis or a dual pathology.
Featured Products for ATTR-CM Diagnostics
Alta DiagnoTech's specialized in vitro diagnostics (IVD) portfolio enables a streamlined diagnostic journey for transthyretin amyloid cardiomyopathy (ATTR-CM), delivering critical insights from initial clinical suspicion through definitive subtyping. If you have related needs, please feel free to contact us for more information or product support.
| Product Name |
Technology |
Application |
| NT-proBNP Quantitative Assay |
Electrochemiluminescence Immunoassay (ECLIA) |
Heart failure assessment and monitoring in ATTR-CM |
| High-Sensitivity Cardiac Troponin I Assay |
Chemiluminescence Immunoassay (CLIA) |
Detection of myocardial injury in cardiacamyloidosis |
| Serum Free Light Chains (FLC) Assay |
Immunoturbidimetric Assay |
Differential diagnosis to rule out AL amyloidosis |
| TTR Genetic Testing Panel |
Next-Generation Sequencing (NGS) |
Detection of TTR gene mutations for hereditaryATTR-CM diagnosis |
| Prealbumin (Transthyretin) Immunoassay |
Chemiluminescence Immunoassay (CLIA) |
Quantification of transthyretin levels in serum |
| Cardiac Function Panel (NT-proBNP + Troponin) |
Multiplex Immunoassay |
Comprehensive assessment of cardiac involvement |
Reference
- Tahara N, Lairez O, Endo J, et al. 99mTechnetium‐pyrophosphate scintigraphy: a practical guide for early diagnosis of transthyretin amyloid cardiomyopathy[J]. ESC Heart Failure, 2022, 9(1): 251-262.
This article is for research use only. Do not use in any diagnostic or therapeutic application.
