- Home
- Resource
- Disease Diagnosis
- Endocrine Diseases
- Diagnosing Congenital Adrenal Hyperplasia: From Newborn Screening to Genetic Confirmation
- Home
- IVD
- By Technology Types
- By Diseases Types
- By Product Types
- Research
- Resource
- Distributors
- Company

Congenital adrenal hyperplasia (CAH) is a group of inherited disorders characterized by enzyme deficiencies in cortisol synthesis, leading to a complex hormonal imbalance with potentially life-threatening consequences. This resource provides a definitive guide to its modern diagnostic pathway, detailing the critical steps from the initial public health safeguard of newborn screening to the essential biochemical confirmation of diagnosis and subtype classification, and finally to the precision of genetic confirmation.
Congenital adrenal hyperplasia (CAH) is a group of inherited autosomal recessive disorders characterized by enzyme deficiencies in the cortisol biosynthesis pathway, most commonly due to mutations in the CYP21A2 gene encoding 21-hydroxylase. This impairment leads to cortisol insufficiency, a compensatory overproduction of adrenocorticotropic hormone (ACTH), and consequent adrenal gland hyperplasia. The resulting hormonal imbalance causes an accumulation of precursor steroids, particularly 17-hydroxyprogesterone (17-OHP), and excessive androgen production. Clinically, CAH presents on a broad spectrum: from severe, life-threatening salt-wasting forms manifesting in newborns with adrenal crisis and ambiguous genitalia (in females), to milder non-classical forms diagnosed later in life with symptoms such as precocious puberty or hirsutism. Early and accurate diagnosis is therefore critical to initiate hormone replacement therapy, prevent acute adrenal insufficiency, manage long-term complications, and provide appropriate genetic counseling.
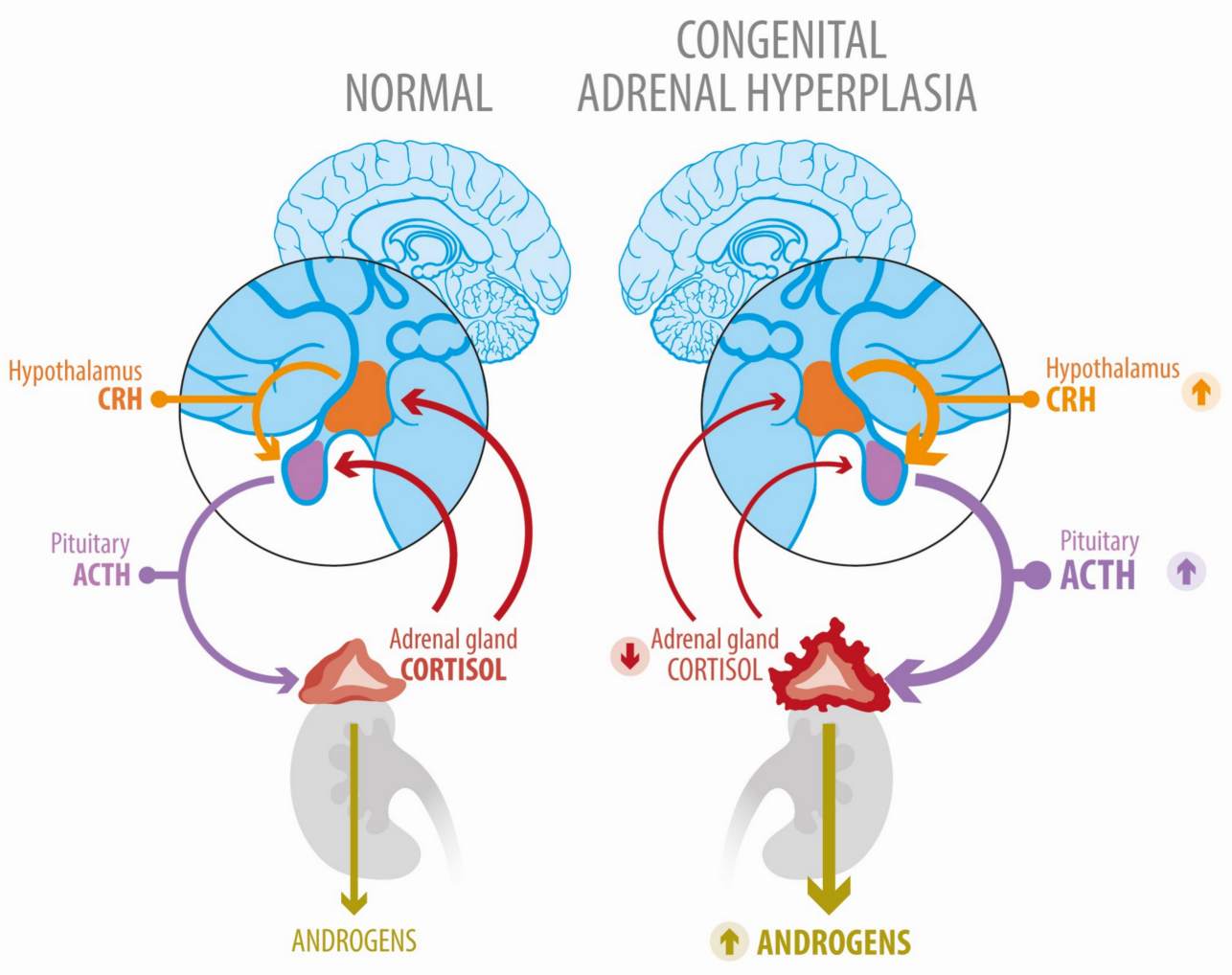 Fig.1 Deregulated hypothalamic–pituitary–adrenal axis function in congenital adrenal hyperplasia due to 21-hydroxylase deficiency. (Uslar T, et al., 2023)
Fig.1 Deregulated hypothalamic–pituitary–adrenal axis function in congenital adrenal hyperplasia due to 21-hydroxylase deficiency. (Uslar T, et al., 2023)
Newborn screening (NBS) for CAH serves as an essential public health intervention and the vital first step in the diagnostic cascade. By detecting the disorder in the first few days of life—before the onset of devastating symptoms like adrenal crisis—NBS allows for the immediate initiation of life-saving glucocorticoid and mineralocorticoid therapy. This proactive intervention prevents severe dehydration, shock, and death in infants with the classic salt-wasting form, while also enabling early management of androgen excess in all affected newborns, thereby mitigating long-term health consequences and improving overall outcomes.

17-OHP is a hallmark steroid hormone of CAH, accumulating due to a deficiency of 21-hydroxylase. While elevated 17-OHP levels in dried blood spots strongly suggest classic CAH, their interpretation is complex. Physiologically elevated 17-OHP levels in preterm infants, low birth weight infants, or sick newborns often lead to false-positive results. To improve accuracy, modern screening protocols employ cutoff values adjusted for birth weight and gestational age, significantly improving the specificity of case identification.

Newborn screening for CAH follows a standardized procedure: within 48 hours of birth, capillary blood is collected via heel prick and deposited onto a filter paper card. The resulting dried blood spot (DBS) is analyzed using a high-throughput automated immunoassay specifically designed for this matrix to quantify 17-hydroxyprogesterone (17-OHP). If the 17-OHP level exceeds a validated, age-corrected threshold, the screening result is positive, requiring immediate diagnostic evaluation.
A positive newborn screen or clinical suspicion of CAH must be followed by definitive biochemical confirmation and precise classification. This critical phase moves from population-based screening to individualized patient diagnosis, utilizing a panel of serum biomarkers and, when necessary, dynamic functional testing. The goal is to establish the diagnosis unequivocally, distinguish between classic salt-wasting, classic simple-virilizing, and non-classical forms, and guide immediate and long-term therapeutic management.

Acute Hormonal Profiling
Following a positive screen, definitive diagnosis relies on serum hormone analysis. The key biomarker is 17-Hydroxyprogesterone (17-OHP); levels in the thousands of ng/dL are diagnostic for classic 21-hydroxylase deficiency. Androstenedione and testosterone are simultaneously measured to quantify androgen excess. To classify the subtype, electrolytes, plasma renin activity, and aldosterone are assessed. An elevated renin with low/normal aldosterone confirms salt-wasting CAH, mandating urgent mineralocorticoid therapy.
Dynamic Stimulation Testing
For cases with equivocal baseline results, particularly suspected non-classical CAH, the dynamic ACTH stimulation test is definitive. It measures 17-OHP before and after a cosyntropin injection to assess adrenal reserve. In NCCAH, stimulated 17-OHP rises to a diagnostic level above normal but below classic CAH thresholds. This test is essential for confirming partial enzyme deficiencies in patients with later-onset symptoms.
While biochemical testing diagnoses the hormonal disorder, genetic analysis provides the definitive molecular map of congenital adrenal hyperplasia (CAH). It confirms the diagnosis at the DNA level, identifies the specific pathogenic mutations, and offers invaluable information for genetic counseling, carrier testing, and prenatal diagnosis. This step is crucial for understanding the inherited basis of the disease and for predicting, albeit imperfectly, potential disease severity.
The Role of Genetic Analysis
Genetic analysis is the definitive diagnostic tool, confirming CAH at the molecular level. Its core roles are to: 1) Establish a precise diagnosis and differentiate CAH from phenocopies; 2) Enable accurate family counseling by identifying carriers and informing recurrence risks; and 3) Facilitate prenatal diagnosis. While genotype can help predict disease severity, the correlation is not absolute due to other modifying factors.
Methodologies for Genetic Testing
Testing strategy is guided by clinical need. For classic 21-hydroxylase deficiency, targeted mutation analysis or Sanger sequencing of the CYP21A2 gene is typically the first-line approach. For complex, atypical, or suspected rare forms of CAH, next-generation sequencing (NGS) panels are the preferred method, as they allow for the simultaneous, comprehensive analysis of all relevant steroidogenesis genes.
As a leading provider of in vitro diagnostic (IVD) products, Alta DiagnoTech offers a comprehensive portfolio of IVD products for congenital adrenal hyperplasia (CAH), supporting the entire diagnostic process from newborn screening to genetic confirmation. Our solutions leverage precise and reliable technologies, including automated immunoassays and advanced molecular platforms, to deliver critical data at every decision point. If you have related needs, please feel free to contact us for more information or product support.
| Product Name | Technology | Application |
| 17-OHP Newborn Screening ELISA/CLIA Kit | Enzyme-Linked Immunosorbent Assay (ELISA) or Chemiluminescent Immunoassay (CLIA) for DBS | High-throughput, quantitative measurement of 17-hydroxyprogesterone (17-OHP) from dried blood spots (DBS) for population-based newborn screening. |
| Serum 17-OHP CLIA Kit | Automated Chemiluminescent Immunoassay (CLIA) | Accurate quantification of serum 17-OHP levels for the biochemical confirmation and monitoring of CAH. |
| Androstenedione & Testosterone CLIA Kits | Automated Chemiluminescent Immunoassay (CLIA) | Simultaneous or individual measurement of key androgen markers to assess the degree of androgen excess in suspected or confirmed CAH patients. |
| Renin Activity (PRA) & Aldosterone ELISA/CLIA Kits | Enzyme-Linked Immunosorbent Assay (ELISA) or Chemiluminescent Immunoassay (CLIA) | Assessment of renin-angiotensin-aldosterone system activity to classify CAH subtype (salt-wasting vs. simple virilizing) and guide mineralocorticoid therapy. |
| CYP21A2 Genotyping Assay | Polymerase Chain Reaction (PCR) & Sanger Sequencing / Targeted Mutation Array | Detection of common pathogenic variants and gene conversions in the CYP21A2 gene for the genetic confirmation of 21-hydroxylase deficiency. |
| Steroidogenesis NGS Panel | Next-Generation Sequencing (NGS) | Comprehensive analysis of multiple genes associated with CAH and other steroidogenic disorders for diagnosing atypical cases, rare CAH forms, and complex phenotypes. |
Reference
This article is for research use only. Do not use in any diagnostic or therapeutic application.
|
There is no product in your cart. |
Copyright © 2026 Alta DiagnoTech. All rights reserved.