Definitive Diagnosis of Familial Hypercholesterolemia (FH): Integrating Clinical Criteria, LDL-C Biomarkers, and Genetic Testing
Familial hypercholesterolemia (FH) is a common yet critically underdiagnosed genetic disorder that causes dangerously high cholesterol from birth and leads to premature heart disease. This definitive resource details the integrated diagnostic approach essential for accurately identifying FH, providing a clear guide to combining structured clinical criteria, precise LDL-C biomarker measurement, and confirmatory genetic testing to secure a diagnosis, enable life-saving family screening, and guide targeted treatment.
Overview of Familial Hypercholesterolemia (FH)
Familial hypercholesterolemia (FH) is a common, inherited autosomal dominant disorder and the most frequent monogenic cause of premature atherosclerotic cardiovascular disease (ASCVD). It is characterized by significantly elevated levels of low-density lipoprotein cholesterol (LDL-C) from birth, resulting from mutations in genes involved in LDL clearance, most commonly the LDL receptor (LDLR) gene. This lifelong exposure to high LDL-C leads to accelerated atherosclerosis, with myocardial infarction potentially occurring as early as the third decade of life. A definitive diagnosis integrates clinical criteria (e.g., Dutch Lipid Clinic Network score), measurement of LDL-C biomarkers, and confirmatory genetic testing, which is crucial not only for confirming the index case but also for enabling cost-effective cascade screening of relatives to prevent catastrophic cardiac events.
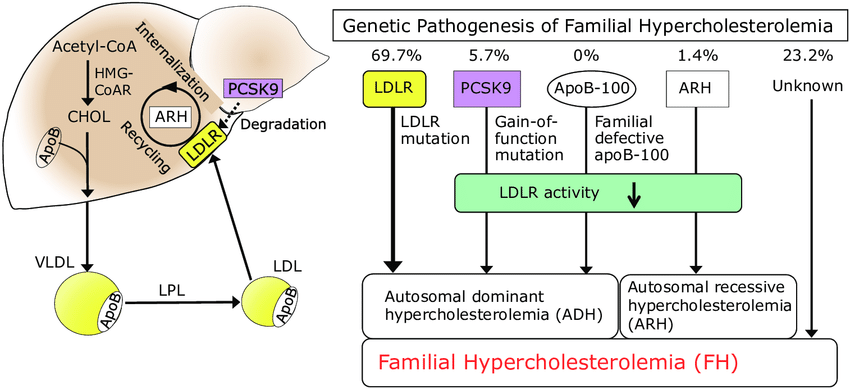 Fig.1 Genetic mechanisms of familial hypercholesterolemia (FH). (Mabuchi, Hiroshi., 2017)
Fig.1 Genetic mechanisms of familial hypercholesterolemia (FH). (Mabuchi, Hiroshi., 2017)
Clinical Criteria – Structured Suspicion and Scoring
Clinical criteria provide a standardized, points-based framework to systematically identify individuals with suspected Familial Hypercholesterolemia (FH) and quantify the likelihood of diagnosis. The most widely accepted system, the Dutch Lipid Clinic Network (DLCN) criteria, integrates personal and family history, physical signs, and laboratory results to generate a score that categorizes suspicion as "Unlikely," "Possible," "Probable," or "Definite" FH. It assigns points for key indicators such as a family history of premature cardiovascular disease or hypercholesterolemia, the patient's own history of premature atherosclerosis, the presence of pathognomonic physical findings like tendon xanthomas, and severely elevated LDL-C levels. This structured approach is essential for the efficient identification of index cases, guiding the decision for confirmatory genetic testing, and initiating cascade screening within families.
LDL-C Biomarkers – The Foundational Laboratory Measurement
The measurement of low-density lipoprotein cholesterol (LDL-C) is the indispensable biochemical cornerstone for diagnosing familial hypercholesterolemia (FH). It serves as the primary, quantifiable biomarker that triggers further diagnostic evaluation and is a key component of all clinical diagnostic criteria. Accurate measurement and correct interpretation of LDL-C levels are fundamental to identifying affected individuals across different age groups.
Diagnostic Thresholds
Diagnosis relies on age-specific LDL-C thresholds. For adults, an untreated LDL-C level greater than 4.9 mmol/L (190 mg/dL) is a primary diagnostic indicator. In children and adolescents, an LDL-C level above 4.0 mmol/L (160 mg/dL) raises strong suspicion, especially with a positive family history. For the rare homozygous FH (HoFH), extremely high untreated levels exceeding 13 mmol/L (500 mg/dL) are characteristic. These thresholds are integrated into clinical scoring systems like the Dutch Lipid Clinic Network criteria.
Critical Nuances
Interpreting LDL-C requires understanding critical nuances. There is significant overlap in levels between FH patients and the general population; a level below the threshold does not rule out FH, particularly in younger individuals or those on treatment. Furthermore, LDL-C is a dynamic biomarker that can be influenced by secondary factors, and triglyceride levels in FH are typically normal. An important adjunct measurement is Lipoprotein(a), as elevated levels significantly modify and increase cardiovascular risk in individuals with FH, independent of their LDL-C.
Genetic Testing – The Confirmatory Gold Standard
Genetic testing represents the definitive, gold-standard method for confirming a diagnosis of familial hypercholesterolemia (FH). It moves beyond phenotypic markers to identify the underlying monogenic cause, providing unequivocal diagnostic clarity. This confirmation is pivotal not only for the index patient but also serves as the cornerstone for effective family-based cascade screening and personalized risk assessment.
Target Genes & Mutations
The primary targets for genetic testing are three core genes. Pathogenic mutations in the LDL receptor (LDLR) gene account for the vast majority (>80%) of FH cases. Approximately 5-10% of cases are caused by mutations in the apolipoprotein B-100 (APOB) gene, and <1% are due to gain-of-function mutations in the PCSK9 gene. It is important to note that in about 20-30% of patients with a clinical FH phenotype, no mutation in these genes is detected, suggesting polygenic or other rare genetic origins.
Clinical Utility
The utility of a positive genetic test is threefold. First, it provides definitive confirmation of the diagnosis, which is crucial for patients with borderline lipid levels or unclear clinical scores. Second, it enables highly efficient and cost-effective cascade screening; once the familial mutation is identified, relatives can be accurately tested for that specific variant. Third, it aids in refined risk stratification, as the specific type of mutation (e.g., null vs. defective LDLR allele) is associated with the severity of LDL-C elevation and future cardiovascular risk.
IVD Products for Familial Hypercholesterolemia (FH)
Alta DiagnoTech provides a comprehensive portfolio of in vitro diagnostic (IVD) solutions for familial hypercholesterolemia (FH), supporting the complete diagnostic pathway from initial lipid screening to definitive genetic confirmation. Our precise and reliable assays empower clinical laboratories to identify at-risk individuals, apply structured clinical criteria with confidence, and implement effective cascade screening programs, thereby addressing the critical challenge of FH underdiagnosis. If you have related needs, please feel free to contact us for more information or product support.
| Product Name |
Technology |
Application |
| Direct LDL-C Quantitative Assay |
Enzymatic / Homogeneous Assay |
Accurate, direct measurement of low-density lipoprotein cholesterol (LDL-C) in serum/plasma, essential for initial screening and applying diagnostic thresholds per clinical criteria (e.g., DLCN). |
| Lipoprotein(a) [Lp(a)] Quantification Kit |
Immunoturbidimetry |
Quantitative measurement of Lipoprotein(a) levels to aid in cardiovascular risk stratification for diagnosed FH patients, as elevated Lp(a) is an independent risk modifier. |
| FH Genetic Screening Panel (LDLR, APOB, PCSK9) |
Next-Generation Sequencing (NGS) Panel |
Comprehensive analysis of the three core genes (LDLR, APOB, PCSK9) to identify pathogenic mutations for confirmatory diagnosis and to establish the familial variant for cascade screening. |
| Targeted FH Mutation Detection Kit |
Real-Time PCR with Melt Curve Analysis |
Rapid, cost-effective genotyping for a curated set of the most common region-specific pathogenic variants in LDLR and APOB, ideal for high-throughput initial genetic screening. |
| LDLR Gene Copy Number Variation (CNV) Assay |
Multiplex Ligation-dependent Probe Amplification (MLPA) |
Detection of large deletions or duplications within the LDLR gene, which are missed by sequencing-based methods but account for a significant portion of mutations. |
Reference
- Mabuchi, Hiroshi. "Half a century tales of familial hypercholesterolemia (FH) in Japan." Journal of atherosclerosis and thrombosis 24.3 (2017): 189-207.
This article is for research use only. Do not use in any diagnostic or therapeutic application.