Pompe disease is a rare but treatable lysosomal disorder that requires urgent and accurate diagnosis to prevent irreversible neuromuscular damage. This resource explores modern diagnostic pathways, from identifying early clinical red flags to implementing cutting-edge biochemical and genetic testing, while addressing key challenges such as pseudodeficiency interference and diagnostic delays.
Overview of Pompe Disease
Pompe disease is a rare, inherited lysosomal storage disorder caused by deficiency of the acid alpha-glucosidase (GAA) enzyme, leading to pathological glycogen accumulation in muscles and nerves. It manifests as a spectrum of severity, from severe infantile-onset Pompe disease (IOPD) to late-onset Pompe disease (LOPD). Early and accurate diagnosis is critical, as delayed intervention results in irreversible damage.
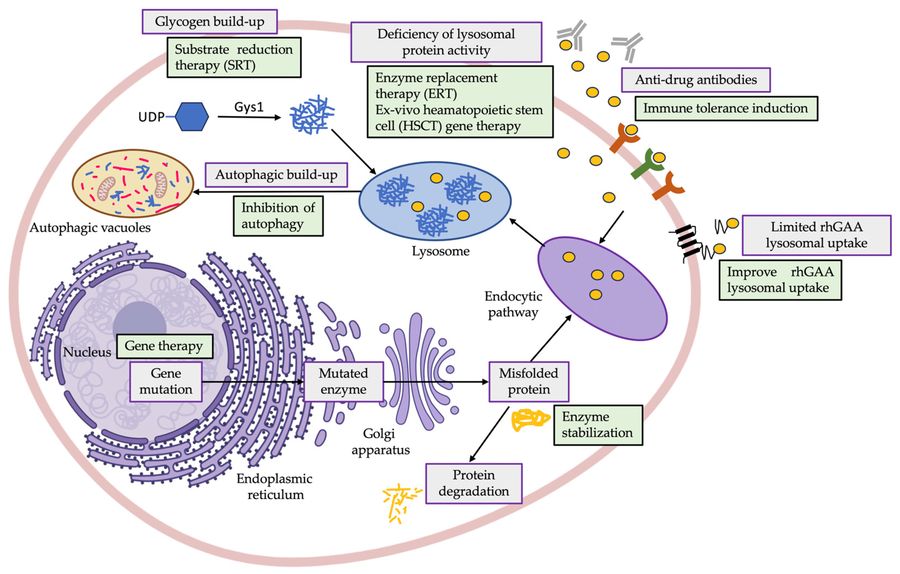 Fig.1 Graphical representation of the most common dysregulations observed in Pompe disease patients. (Gómez-Cebrián N, et al., 2023)
Fig.1 Graphical representation of the most common dysregulations observed in Pompe disease patients. (Gómez-Cebrián N, et al., 2023)
Diagnosis of Pompe Disease
Diagnostic approaches for Pompe disease include enzyme activity assays (gold standard) and genetic testing. The primary screening method is a dried blood spot (DBS) test that measures acid alpha-glucosidase (GAA) activity, supplemented by leukocyte/fibroblast testing to reduce false positives due to pseudo-deficient alleles. GAA gene sequencing is essential for confirming the diagnosis, classifying variants, and predicting disease severity. However, challenges remain, including delays in diagnosis, differences in GAA cutoffs across regions, and limited global standardization of newborn screening (NBS).
Diagnostic Workflow for Pompe Disease
Pompe disease diagnosis requires a stepwise approach integrating clinical evaluation, biochemical testing, genetic confirmation, and biomarker analysis to ensure accuracy and guide timely treatment. Due to its variable presentation, from severe infantile-onset cases to milder late-onset forms, the diagnostic pathway must address both urgent detection and chronic disease identification. Below, we detail the four critical phases:
Initial Clinical Suspicion
Diagnosis begins by recognizing subtype-specific red flags: infantile-onset cases present with hypotonia, cardiomyopathy, and failure to thrive, while late-onset disease manifests as progressive muscle weakness and respiratory decline. Clinicians should suspect Pompe when encountering unexplained myopathy or delayed milestones, especially with elevated CK levels.
First-Line Biochemical Testing
The diagnosis of Pompe disease begins with GAA enzyme activity measurement, typically using dried blood spots (DBS) or leukocytes. DBS offers practical screening with fluorometric/MS-MS methods (cutoff: <1-3% activity), while leukocyte assays provide higher specificity by avoiding pseudodeficiency interference.
Confirmatory Genetic Testing
Definitive diagnosis requires GAA gene sequencing to identify pathogenic variants and distinguish true Pompe disease from benign pseudodeficiency. This step confirms diagnosis, predicts disease severity (infantile vs. late-onset), and enables family counseling and therapy selection.
Supporting Biomarkers
While nonspecific, urinary Glc4 and elevated CK levels support diagnosis and monitor treatment response. These biomarkers complement enzyme/genetic testing, particularly in ambiguous cases or for tracking disease progression.
Challenges in Pompe Disease Diagnosis
Diagnosing Pompe disease remains complex due to heterogeneous symptoms, technical limitations of testing, and systemic healthcare gaps. These challenges often delay diagnosis, particularly for late-onset cases, leading to irreversible progression before treatment begins.
- Clinical Recognition Gaps: Pompe disease symptoms often mimic other neuromuscular disorders, leading to frequent misdiagnosis and delayed identification.
- Limitations of Biochemical Testing: False positives/negatives in GAA enzyme assays arise from pseudodeficiency alleles and sample stability issues.
- Genetic Interpretation Complexities: Variants of uncertain significance (VUS) and large deletions in the GAA gene complicate definitive diagnosis.
- Systemic and Access Barriers: Limited newborn screening programs and high diagnostic costs restrict testing availability, especially in low-resource regions.
- Post-Diagnostic Challenges: Lack of standardized biomarkers makes treatment monitoring and disease progression tracking difficult.
Future of Pompe Disease Diagnosis
The diagnosis of Pompe disease is evolving toward faster, more accessible, and precise methods, driven by advances in high-throughput newborn screening (NBS) with tandem mass spectrometry (MS/MS), AI-assisted genetic variant interpretation, and point-of-care enzyme testing. Emerging technologies like wearable biosensors for real-time biomarker monitoring promise to reduce diagnostic delays, particularly for late-onset cases. Global efforts are also addressing equity gaps through cost-effective dried blood spot (DBS) protocols and multiplexed lysosomal enzyme panels that screen for Pompe alongside other metabolic disorders.
Alta DiagnoTech provides cutting-edge IVD solutions for Pompe disease, offering a highly accurate GAA enzyme test kit, advanced genetic testing panels, and innovative biomarker testing tools. If you have related needs, please feel free to contact us for more information or product support.
Reference
- Gómez-Cebrián N, Gras-Colomer E, Poveda Andrés J L, et al. Omics-based approaches for the characterization of pompe disease metabolic phenotypes[J]. Biology, 2023, 12(9): 1159.
This article is for research use only. Do not use in any diagnostic or therapeutic application.