- Home
- Resource
- Disease Diagnosis
- Genetic Diseases
- Navigating Wilson Disease Diagnosis: Biomarkers, Genetics, and Actionable Algorithms
- Home
- IVD
- By Technology Types
- By Diseases Types
- By Product Types
- Research
- Resource
- Distributors
- Company

Wilson disease (WD), a rare but treatable genetic disorder of copper metabolism, poses significant diagnostic challenges due to its highly variable clinical presentations and limitations of traditional biomarkers. This comprehensive resource provides healthcare professionals with a step-by-step guide to modern WD diagnosis, covering essential biomarkers, genetic testing and actionable diagnostic algorithms.
Wilson disease is a rare, inherited disorder of copper metabolism caused by mutations in the ATP7B gene, leading to toxic copper accumulation in the liver, brain, and other organs. Characterized by highly variable clinical presentations, including hepatic disease, neurological symptoms (e.g., tremors, dystonia), and psychiatric manifestations, Wilson disease demands early diagnosis to prevent irreversible organ damage. Timely detection hinges on a combination of biochemical tests and genetic analysis, yet diagnostic delays remain common due to symptom overlap with other conditions.
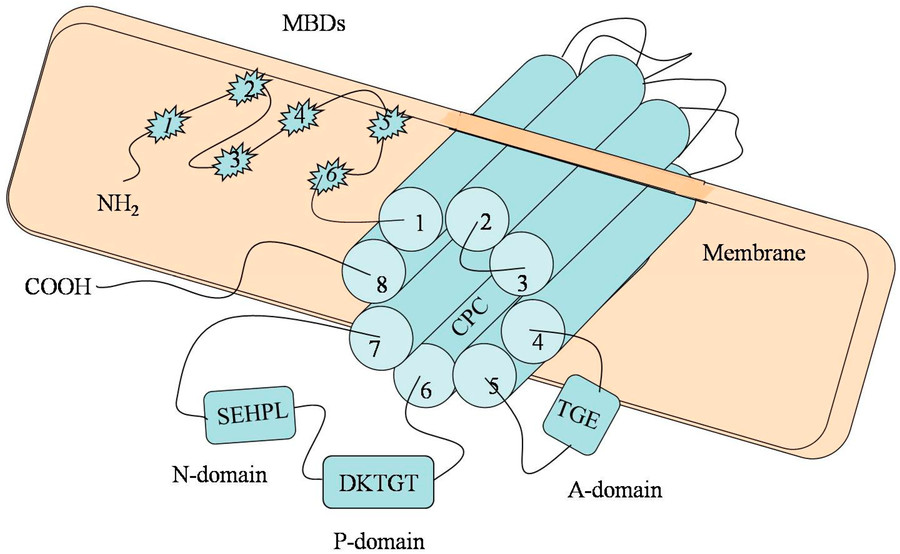 Fig.1 Schematic representation of the ATP7B gene. (Wu F, et al., 2015)
Fig.1 Schematic representation of the ATP7B gene. (Wu F, et al., 2015)
Accurate diagnosis of Wilson disease hinges on the interpretation of key biochemical markers that reflect copper metabolism dysfunction. These biomarkers not only support clinical suspicion but also help differentiate Wilson disease from other hepatic or neurological disorders. The cornerstone tests include ceruloplasmin levels, 24-hour urinary copper excretion, and hepatic copper quantification.
Ceruloplasmin, the primary copper-transporting protein, is typically reduced (<0.2 g/L) in Wilson disease due to impaired incorporation of copper into the protein. However, false negatives occur in 20% of cases, particularly in acute inflammation or during pregnancy, where ceruloplasmin may rise into the normal range.
This non-invasive test measures copper excretion in urine, with levels >100 μg/24 hours strongly supporting WD. It is particularly useful in symptomatic patients, where elevated urinary copper reflects systemic copper overload. However, proper collection is crucial—contamination or incomplete sampling can skew results.
Liver biopsy with copper quantification (>250 μg/g dry weight) remains the gold standard for Wilson disease diagnosis, especially in ambiguous cases. It is highly specific but invasive, and sampling error can occur due to uneven copper distribution in cirrhosis. This test is most valuable when other biomarkers are inconclusive.
Genetic testing plays a key role in confirming the diagnosis of Wilson disease, especially in cases with equivocal biomarker results. By identifying the causative mutation in the ATP7B gene, genetic analysis can provide a definitive diagnosis and support family screening. With more than 600 mutations reported, traditional Sanger sequencing is complex and requires comprehensive approaches such as next-generation sequencing (NGS) to capture both known and novel variants.
When to Use Genetic Testing
Genetic testing for Wilson disease is recommended in three key scenarios: (1) confirmatory diagnosis for patients with suggestive symptoms but inconclusive biochemical results, (2) family screening of first-degree relatives to identify asymptomatic carriers or affected individuals, and (3) prenatal/preconception counseling for at-risk couples.

Testing Methodologies
The choice of genetic test depends on clinical context and population: targeted mutation analysis offers a cost-effective first step, while comprehensive NGS panels detect rare or novel ATP7B mutations across diverse populations. Deletion/duplication analysis (e.g., MLPA) complements sequencing to identify large genomic rearrangements, ensuring complete coverage for diagnostic certainty.

Interpretation Challenges
Genetic results require careful clinical correlation due to: (1) variants of uncertain significance (VUS), which necessitate biochemical/clinical follow-up, (2) compound heterozygosity (two different mutations), which may obscure phenotype predictions, and (3) incomplete penetrance, where some mutation carriers remain asymptomatic. Integration with Leipzig Score criteria helps standardize diagnostic decisions.
A structured diagnostic algorithm is essential to navigate the complexities of Wilson disease (WD), integrating clinical suspicion, biomarker results, and genetic testing into a stepwise workflow. Initial evaluation typically begins with ceruloplasmin and 24-hour urinary copper for high-sensitivity screening, followed by slit-lamp examination for Kayser-Fleischer rings in neurological cases. Ambiguous results trigger genetic testing (ATP7B sequencing) or liver biopsy for copper quantification, while the Leipzig Score provides a standardized diagnostic framework. This approach minimizes delays, especially for atypical presentations, and ensures timely intervention.
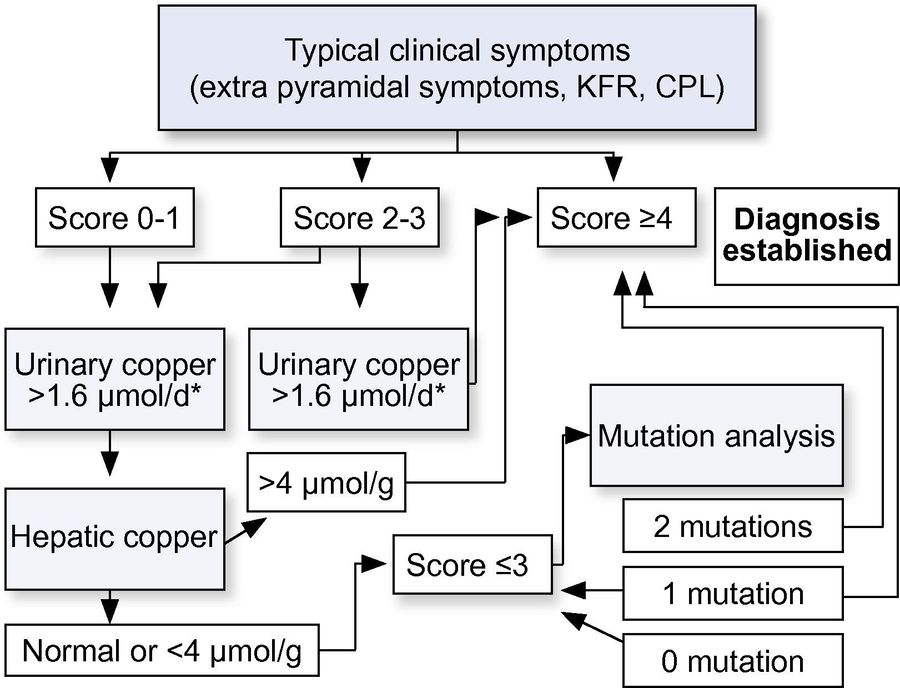 Fig.2 Diagnostic algorithms for Wilson's disease based on the Leipzig Score. (European Association for the Study of the Liver, 2012)
Fig.2 Diagnostic algorithms for Wilson's disease based on the Leipzig Score. (European Association for the Study of the Liver, 2012)
The diagnostic landscape for Wilson disease is poised for transformation through noninvasive biomarkers (e.g., serum exchangeable copper, microRNAs), AI-powered interpretation tools to integrate multi-omics data, and rapid point-of-care genetic tests enabling faster ATP7B variant detection. Emerging technologies like biosensors and advanced imaging (e.g., hepatic copper MRI quantification) could further reduce reliance on invasive biopsies. These innovations aim to address current gaps, particularly in pediatric and resource-limited settings, by delivering earlier, more accurate, and accessible diagnosis, ultimately improving outcomes for this treatable but often missed disorder.
Alta DiagnoTech provides cutting-edge in vitro diagnostic (IVD) solutions for Wilson disease, offering high-precision test kits that streamline the detection of key biomarkers, including ceruloplasmin, urinary copper, and hepatic copper. If you have related needs, please feel free to contact us for more information or product support.
References
This article is for research use only. Do not use in any diagnostic or therapeutic application.
|
There is no product in your cart. |
Copyright © 2026 Alta DiagnoTech. All rights reserved.