- Home
- Resource
- Disease Diagnosis
- Genetic Diseases
- Stop Missing Fabry Disease: Advanced Diagnostic Pathways for Clinicians and Labs
- Home
- IVD
- By Technology Types
- By Diseases Types
- By Product Types
- Research
- Resource
- Distributors
- Company

Fabry disease (FD) is a multifaceted genetic disorder that demands comprehensive diagnostic strategies to overcome its diagnostic challenges. This resource provides an in-depth exploration of current and emerging diagnostic approaches, including enzymatic assays, biomarker testing, and genetic analysis, while addressing critical issues like gender disparities and organ-specific manifestations.
Fabry disease (FD) is a rare, X-linked lysosomal storage disorder caused by mutations in the GLA gene, leading to deficient activity of the enzyme α-galactosidase A (α-Gal A). This results in progressive accumulation of globotriaosylceramide (Gb3) and its derivatives in cells, damaging vital organs such as the kidneys, heart, and nervous system. FD affects an estimated 1 in 40,000 to 1 in 117,000 live births worldwide, though recent newborn screening studies suggest it may be significantly underdiagnosed, with higher prevalence in certain populations.
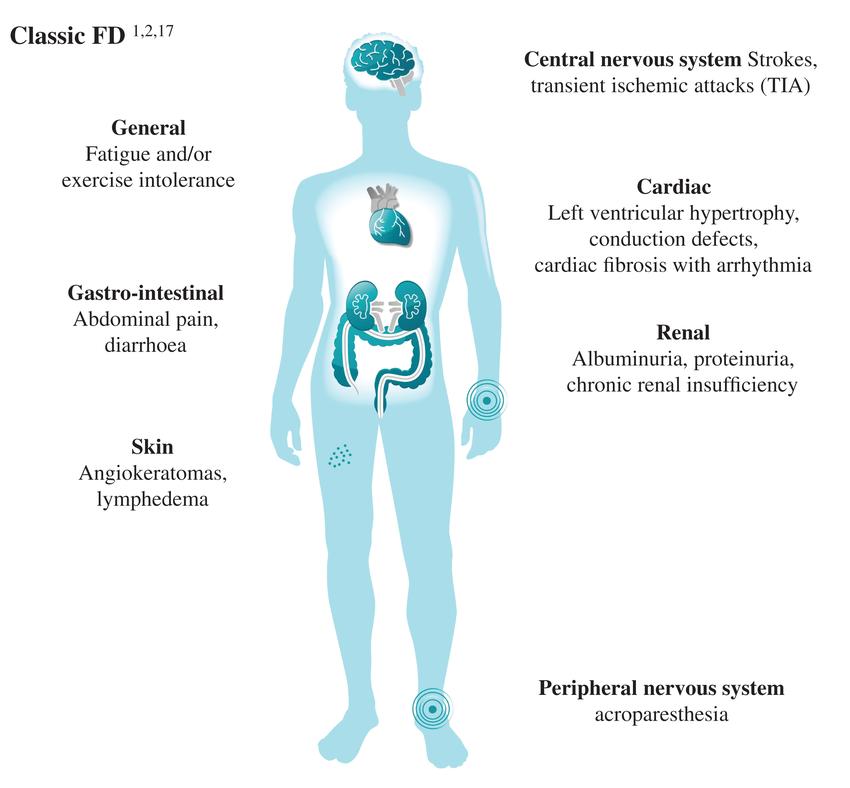 Fig.1 Main clinical manifestations of Fabry disease (FD). (Germain D P, et al., 2022)
Fig.1 Main clinical manifestations of Fabry disease (FD). (Germain D P, et al., 2022)
Fabry disease (FD) diagnosis integrates biochemical, genetic, and clinical assessments due to its heterogeneous presentation. In males, reduced α-galactosidase A (α-Gal A) enzyme activity in leukocytes or dried blood spots provides initial confirmation, while females often require GLA gene sequencing due to normal enzyme levels. The biomarker lyso-Gb3 enhances diagnostic accuracy, particularly for atypical cases.
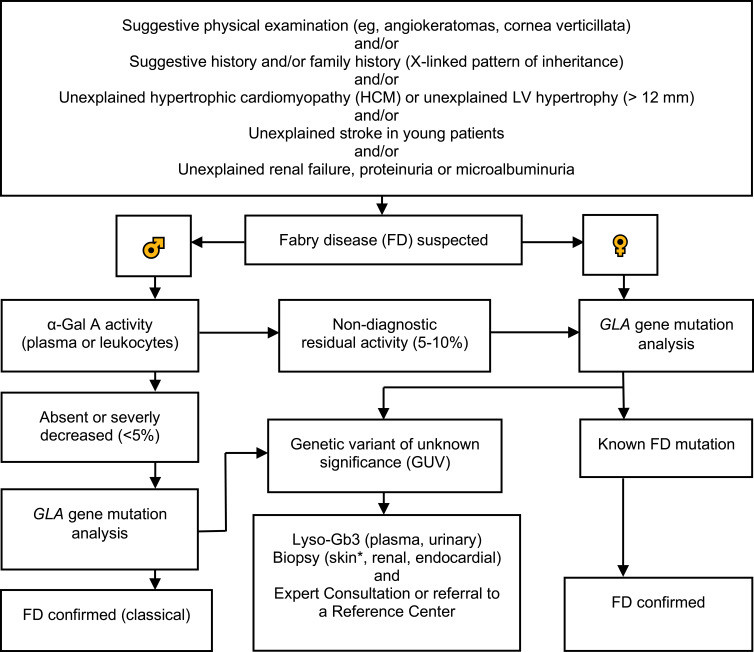 Fig.2 Diagnostic algorithm for Fabry disease (FD). (Vardarli I, et al., 2020)
Fig.2 Diagnostic algorithm for Fabry disease (FD). (Vardarli I, et al., 2020)
Cardiac complications are a leading cause of morbidity and mortality in Fabry disease (FD), affecting up to 60-70% of patients, including both classic and late-onset variants. The spectrum of cardiac manifestations includes left ventricular hypertrophy (LVH), arrhythmias, valvular dysfunction, and heart failure, often mimicking hypertrophic cardiomyopathy (HCM) or other storage disorders. The key diagnostic methods are as follows:
Cardiac imaging is pivotal for detecting Fabry-related cardiomyopathy. Echocardiography reveals concentric left ventricular hypertrophy (LVH), while cardiac MRI (CMR) provides superior tissue characterization through late gadolinium enhancement (fibrosis) and quantitative T1 mapping (lipid accumulation). CMR is particularly valuable for early detection before hypertrophy develops.
ECG abnormalities in FD often precede structural changes, with characteristic findings including short PR intervals (50% of cases), conduction defects, and arrhythmias. Progressive ECG changes correlate with disease severity, making serial monitoring crucial for risk stratification.

Plasma globotriaosylsphingosine (Lyso-Gb3) levels strongly associate with cardiac involvement severity. Cardiac-specific biomarkers (high-sensitivity troponin, NT-proBNP) complement imaging by detecting subclinical myocardial injury and monitoring treatment response.

GLA gene sequencing is mandatory for confirming FD in patients with unexplained LVH, especially women and late-onset cases. Genetic testing enables family screening and differentiation from other hypertrophic cardiomyopathies, with certain variants predicting severe cardiac phenotypes.
Renal complications in Fabry disease (FD) arise from globotriaosylceramide (Gb3) accumulation in podocytes, tubular cells, and vascular endothelium, leading to progressive kidney damage. Early diagnosis is critical to prevent end-stage renal disease (ESRD), which affects ~13% of male and ~2% of female patients by age 55. Some key diagnostic methods are listed below:
Neurologic manifestations in Fabry disease (FD) include small-fiber neuropathy, cerebral white matter lesions, and stroke-like episodes, often presenting before systemic complications. Diagnosis relies on clinical assessment, quantitative sensory testing, skin biopsy, and brain MRI. Plasma lyso-Gb3 levels may correlate with neurologic severity, while neurophysiologic studies (e.g., nerve conduction) often remain normal due to selective small-fiber damage. Early recognition is critical to prevent irreversible neurologic damage through timely enzyme replacement therapy.
Fabry disease (FD) remains a frequently underdiagnosed and misdiagnosed X-linked lysosomal storage disorder due to its heterogeneous clinical presentation, which often mimics more common conditions such as chronic kidney disease, hypertrophic cardiomyopathy, or neuropathic pain syndromes. The diagnostic delay averages 13.7 years in males and 16.3 years in females, with many patients visiting multiple specialists before receiving a correct diagnosis9. Key challenges include:
The future of Fabry disease (FD) diagnosis lies in integrating advanced technologies to overcome current challenges. Next-generation sequencing (NGS) and AI-powered variant interpretation will improve genetic screening accuracy, while high-sensitivity Lyso-Gb3 assays and multi-omics profiling may enable earlier detection. Point-of-care testing (POCT) and digital health tools could reduce diagnostic delays in primary care.
Alta DiagnoTech provides comprehensive IVD solutions for Fabry disease (FD) diagnostics, including reagents, kits and devices related to enzyme activity assays, biomarker testing and genetic screening, designed to enable accurate, early detection and personalized disease management. If you have related needs, please feel free to contact us for more information or product support.
References
This article is for research use only. Do not use in any diagnostic or therapeutic application.
|
There is no product in your cart. |
Copyright © 2026 Alta DiagnoTech. All rights reserved.