Gaucher disease, a rare inherited lysosomal disorder caused by GBA1 mutations, requires accurate and timely diagnosis to enable life-changing interventions. This resource provides a thorough overview of modern diagnostic approaches, from gold-standard enzyme activity assays and disease-specific biomarkers (Lyso-Gb1, chitotriosidase) to advanced genetic analysis.
Introduction to Gaucher Disease
Gaucher disease is a rare, inherited lysosomal storage disorder caused by deficient activity of the enzyme glucocerebrosidase (GCase) due to mutations in the GBA1 gene. This leads to toxic accumulation of glucosylceramide and glucosylsphingosine (Lyso-Gb1) in macrophages, primarily affecting the liver, spleen, bone marrow, and the central nervous system. GD manifests in three subtypes:
| Subtypes |
Common Mutations |
Key Symptoms |
Prevalence |
| Type 1 (Non-neuronopathic) |
N370S |
Hepatosplenomegaly, cytopenia (anemia/thrombocytopenia), bone pain/fractures, no neurological involvement |
~95% |
| Type 2 (Acute neuronopathic) |
L444P, RecNciI |
Rapid neurodegeneration (seizures, hypertonia), oculomotor apraxia, failure to thrive, hepatosplenomegaly |
~1% |
| Type 3 (Chronic neuronopathic) |
L444P, D409H |
Slower neurological progression (ataxia, myoclonus), hepatosplenomegaly, ophthalmologic abnormalities, cardiac valve calcification (subtype 3c) |
~5% |
Diagnosis of Gaucher Disease
The accurate diagnosis of Gaucher disease (GD) is critical to enable timely intervention with enzyme replacement therapy (ERT) or substrate reduction therapy (SRT), preventing irreversible organ damage and improving quality of life. However, diagnosis faces significant challenges, including underdiagnosis due to nonspecific symptoms, phenotypic variability, and limitations in differentiating neuronopathic subtypes early in disease progression. Additionally, pseudodeficiency alleles complicate genetic interpretation, while access to specialized testing remains limited in resource-poor settings.
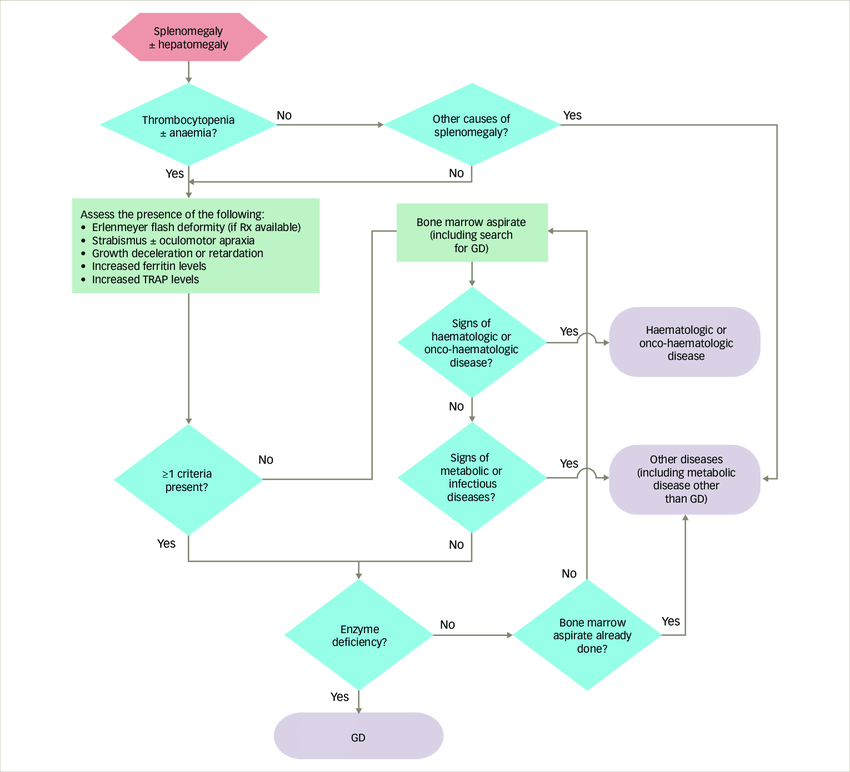 Fig.1 Algorithm for the diagnosis of Gaucher disease in the paediatric population. (Cappellini M D, et al., 2018)
Fig.1 Algorithm for the diagnosis of Gaucher disease in the paediatric population. (Cappellini M D, et al., 2018)
Enzyme Activity Assays
Enzyme activity assays are the gold standard for diagnosing Gaucher disease (GD), directly measuring β-glucocerebrosidase (GCase) activity in patient samples to confirm functional deficiency. The main methods for enzyme activity detection are as follows:
Leukocyte-Based Assays
The gold-standard method measures β-glucocerebrosidase (GCase) activity in isolated white blood cells using fluorogenic substrates (e.g., 4-MUG). While highly accurate for diagnosing Gaucher disease (activity typically <15% of normal), it requires fresh blood samples and specialized processing, and may yield ambiguous results with pseudodeficiency variants (e.g., E326K).
Dried Blood Spot (DBS) Assays
This high-throughput screening method analyzes GCase activity from dried blood spots on filter paper, enabling population-wide newborn screening and testing in resource-limited settings. Though convenient and stable at room temperature, DBS assays show slightly reduced sensitivity compared to leukocyte methods and require confirmatory testing for definitive diagnosis.
Fluorometric/MS-MS Assays
Advanced platforms like tandem mass spectrometry (MS/MS) provide superior precision by simultaneously quantifying GCase activity and pathogenic biomarkers (e.g., Lyso-Gb1). These methods are increasingly adopted for their high sensitivity, ability to detect multiple analytes, and compatibility with automated systems, though they require specialized instrumentation and expertise.
Biomarker Testing
Biomarker testing plays a pivotal role in diagnosing Gaucher disease (GD), monitoring disease progression, and evaluating treatment response. These quantitative assays complement enzyme activity and genetic testing by providing real-time insights into pathological substrate accumulation and disease severity.
Glucosylsphingosine (Lyso-Gb1)
Lyso-Gb1, the most specific biomarker for Gaucher disease, accumulates due to glucocerebrosidase deficiency. Quantified via LC-MS/MS, it demonstrates 100-1000x elevation in GD patients versus controls, strongly correlating with disease severity and treatment response.
Chitotriosidase
This macrophage-derived enzyme, elevated in ~90% of GD patients, has been a traditional biomarker for two decades. While useful for monitoring disease activity, its utility is limited by 5-6% false negatives and non-specific elevation in other inflammatory conditions.
CCL18/PARC
A complementary macrophage activation marker, CCL18 serves as an alternative when chitotriosidase is deficient. Though less specific than Lyso-Gb1, it provides additional clinical information, particularly in assessing therapeutic response and disease progression.
Genetic Analysis
Genetic testing is essential for confirming Gaucher disease (GD) diagnosis, predicting disease severity, and guiding family counseling. The primary target is the GBA1 gene, where over 400 pathogenic variants have been identified.
- Common Pathogenic Variants: The GBA1 gene contains several recurrent pathogenic variants associated with Gaucher disease subtypes. The N370S variant (c.1226A>G) is most common in type 1 Gaucher disease, while L444P (c.1448T>C) and recNciI are associated with a severe neuropathy type (type 2/3). Other variants, such as 84GG and IVS2+1G>A, are associated with early-onset disease.
- Testing Methods: Genetic analysis employs Sanger sequencing for targeted variant detection and NGS panels for comprehensive GBA1 screening, including pseudogene discrimination. MLPA identifies large deletions/duplications, while emerging long-read sequencing resolves complex structural variants. These methods require integration with enzyme/biomarker data to distinguish pathogenic mutations from benign polymorphisms or pseudodeficiency variants.
Conclusion
The diagnosis of Gaucher disease (GD) demands a multidisciplinary strategy, combining enzyme activity assays, biomarker quantification, and genetic analysis to overcome diagnostic challenges and enable precision management. While glucocerebrosidase (GCase) testing remains the gold standard, emerging tools like Lyso-Gb1 biomarkers and NGS-based GBA1 sequencing are transforming detection accuracy, particularly for atypical cases and therapeutic monitoring.
Alta DiagnoTech delivers trusted IVD solutions for Gaucher disease (GD), offering high-performance diagnostic products with demonstrated advantages in accuracy, rapid turnaround time, and seamless integration into clinical workflows. If you have related needs, please feel free to contact us for more information or product support.
Reference
- Cappellini M D, Cassinerio E, Motta I, et al. Finding and treating Gaucher disease type 1–The role of the haematologist[J]. Eur Oncol Haematol, 2018, 14(1): 50-56.
This article is for research use only. Do not use in any diagnostic or therapeutic application.