- Home
- Resource
- Disease Diagnosis
- Genetic Diseases
- From Newborn Screening to Genetic Testing: A Complete Diagnostic Guide for Phenylketonuria (PKU)
- Home
- IVD
- By Technology Types
- By Diseases Types
- By Product Types
- Research
- Resource
- Distributors
- Company

Phenylketonuria (PKU), a potentially devastating yet treatable metabolic disorder, demands accurate and timely diagnosis to prevent irreversible neurological damage. This comprehensive resource explores the complete diagnostic journey, from critical newborn screening protocols using tandem mass spectrometry (MS/MS) to advanced confirmatory testing and genetic analysis.
Phenylketonuria (PKU) is an inherited metabolic disorder caused by mutations in the PAH gene, leading to deficient phenylalanine hydroxylase (PAH) enzyme activity and subsequent accumulation of phenylalanine (Phe) in the blood. If left untreated, elevated Phe levels result in severe neurotoxicity, causing intellectual disability, seizures, and behavioral abnormalities. Early diagnosis through newborn screening (NBS) and immediate dietary intervention (low-Phe diet or enzyme therapy) can prevent complications, highlighting the critical role of timely and accurate diagnostic methods.
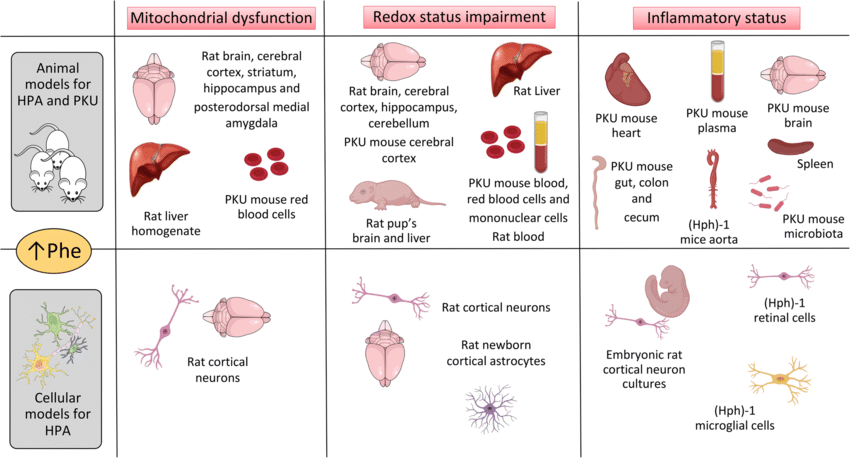 Fig.1 Pathological mechanisms involved in phenylketonuria (PKU) and other conditions with hyperphenylalaninemia (HPA). (Wyse A T S, et al., 2021)
Fig.1 Pathological mechanisms involved in phenylketonuria (PKU) and other conditions with hyperphenylalaninemia (HPA). (Wyse A T S, et al., 2021)
Tandem mass spectrometry (MS/MS) is the gold-standard method for newborn PKU screening, enabling high-throughput, multiplexed analysis of phenylalanine (Phe) and tyrosine (Tyr) from dried blood spots. This technique uses two mass analyzers in sequence: the first isolates Phe/Tyr ions, while the second quantifies them with high specificity, achieving sensitivity down to 50–100 μmol/L and minimizing false positives. MS/MS simultaneously screens for 30+ other metabolic disorders, making it cost-effective for large-scale programs.
Newborn screening (NBS) for phenylketonuria (PKU) is a critical public health initiative that enables early detection and intervention to prevent severe neurological damage and intellectual disability. Implemented in over 60 countries, NBS programs have dramatically reduced PKU-related complications, though coverage gaps persist in low-resource regions.
A positive newborn screening (NBS) result for phenylketonuria (PKU) requires urgent confirmatory testing to establish a definitive diagnosis, exclude false positives, and guide critical treatment decisions. Without confirmation, infants risk either unnecessary dietary restrictions or delayed intervention leading to irreversible neurological damage. This phase combines quantitative biochemical testing, urine analysis, and genetic analysis to accurately classify PKU and rule out mimics like BH4 deficiency.

The gold-standard plasma phenylalanine (Phe) measurement via HPLC or MS/MS provides precise quantification, with levels >360 μmol/L confirming PKU. Concurrent Phe/Tyrosine (Tyr) ratio analysis (typically >2 in PKU) helps distinguish true PKU from transient hyperphenylalaninemia. Repeat testing ensures reliability, as early samples may be influenced by breastfeeding patterns or parenteral nutrition.

In untreated PKU, urine tests detect phenylpyruvate and other phenylketones, producing a characteristic "musty odor." While less specific than blood tests, urine analysis supports diagnosis in symptomatic infants and monitors dietary compliance. It also aids in identifying BH4 deficiency through abnormal pterin metabolites, which requires distinct therapy.

PAH gene sequencing identifies pathogenic variants, confirming PKU and enabling genotype-phenotype correlations (e.g., predicting disease severity). It also facilitates carrier testing for family planning and prenatal diagnosis. Emerging next-generation sequencing (NGS) panels improve detection of rare variants and large deletions.
Confirming phenylketonuria (PKU) requires excluding other conditions that may mimic its biochemical or clinical features, such as benign hyperphenylalaninemia (HPA), tetrahydrobiopterin (BH4) deficiency, or maternal PKU. Distinguishing these conditions is critical, as management differs significantly. Differential diagnosis relies on Phe/Tyr ratios, pterin profiling, dihydrobiopterin reductase (DHPR) activity assays, and genetic testing to ensure accurate classification and targeted therapy.
While current diagnostic methods like MS/MS and genetic testing have revolutionized PKU detection, challenges remain, including false positives/negatives in newborn screening, limited access to confirmatory testing in resource-poor regions, and difficulties in monitoring dietary compliance. The future lies in portable biosensors for real-time Phe tracking, AI-powered interpretation of complex metabolic data, and non-invasive techniques (e.g., saliva-based assays) to improve patient adherence.
Specializing in Phenylketonuria (PKU) diagnostics, Alta DiagnoTech's precise IVD kits enable exact diagnosis and personalized care, facilitating accurate identification from newborn screening throughout lifelong monitoring. If you have related needs, please feel free to contact us for more information or product support.
References
This article is for research use only. Do not use in any diagnostic or therapeutic application.
|
There is no product in your cart. |
Copyright © 2026 Alta DiagnoTech. All rights reserved.