- Home
- Resource
- Disease Diagnosis
- Metabolic Diseases
- Decoding Lysosomal Storage Disorders (LSD): Advanced IVD Strategies for Accurate Diagnosis
- Home
- IVD
- By Technology Types
- By Diseases Types
- By Product Types
- Research
- Resource
- Distributors
- Company

Lysosomal storage disorders (LSDs) represent a complex group of inherited metabolic diseases requiring accurate and timely diagnosis for effective management. This resource provides a complete overview of contemporary diagnostic strategies, from foundational enzyme activity assays to advanced molecular techniques. We explore the clinical utility, advantages, and limitations of each method while highlighting emerging technologies shaping the future of LSD diagnostics.
Lysosomal storage disorders (LSD) are a group of inherited metabolic disorders caused by defects in lysosomal function, resulting in the intracellular accumulation of undegraded substrates, such as complex lipids, glycoproteins, glycosaminoglycans, etc. These diseases affect multiple organ systems, including the brain, liver, spleen, bones, and cardiovascular system, and often lead to progressive and severe clinical manifestations.
Table 1 Mutated genes and deficient enzymes in some lysosomal storage disorders (LSDs).
| Disease Name | Enzyme Deficiency | Mutated Gene | Incidence (Approximate) |
| Pompe Disease | Acid α-glucosidase (GAA) | GAA | 1/40,000 |
| Gaucher Disease | Glucocerebrosidase (GBA) | GBA | 1/40,000-1/60,000 |
| Niemann-Pick Disease | Type A/B: Acid sphingomyelinase (ASM) Type C: NPC1/NPC2 protein dysfunction |
SMPD1 (Type A/B) NPC1/NPC2 (Type C) |
Type A/B: 1/250,000 Type C: 1/100,000 |
| GM2 Gangliosidosis | Tay-Sachs: Hexosaminidase A (HEXA) Sandhoff: Hexosaminidase B (HEXB) |
HEXA (Tay-Sachs) HEXB (Sandhoff) |
Tay-Sachs: 1/3,200 (Ashkenazi Jews) Sandhoff: 1/300,000 (general population) |
Diagnosis of lysosomal storage disorders (LSD) involves a multi-step approach combining clinical suspicion, biochemical testing, and genetic confirmation. Initial screening typically relies on enzyme activity assays to detect deficiencies in lysosomal enzymes. Biomarkers and imaging support differential diagnosis. Genetic testing identifies pathogenic variants in associated genes, aiding in subtype classification and family counseling.
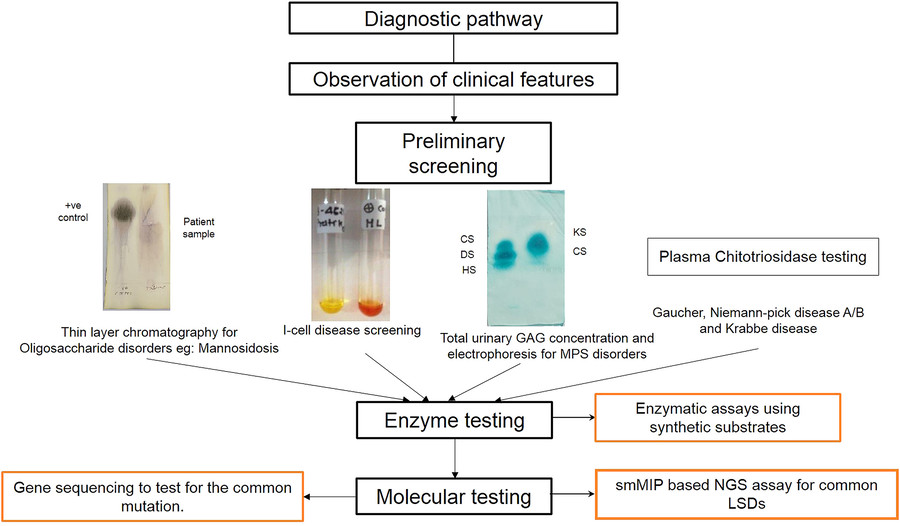 Fig.1 Diagnostic algorithm for lysosomal storage disorders (LSD). (Sheth J, et al., 2023)
Fig.1 Diagnostic algorithm for lysosomal storage disorders (LSD). (Sheth J, et al., 2023)
Accurate measurement of lysosomal enzyme activity is critical for diagnosing lysosomal storage disorders (LSD). Among the various techniques available, fluorometry and tandem mass spectrometry (MS/MS) have emerged as two of the most widely used and reliable methods. Fluorometry offers high sensitivity and simplicity, making it suitable for initial screening, while MS/MS provides superior specificity and multiplexing capabilities, enabling simultaneous analysis of multiple enzymes.

Fluorometry Method
This method relies on synthetic fluorescent substrates that are cleaved by the target lysosomal enzyme, generating a measurable fluorescent signal proportional to enzyme activity. Fluorometry is particularly suitable for high-throughput screening in clinical laboratories, as it requires minimal sample preparation and standard instrumentation (e.g., microplate readers).
Tandem Mass Spectrometry (MS/MS) Method
MS/MS method uses stable isotope-labeled substrates, which are processed by lysosomal enzymes, and the resulting products are quantified by mass spectrometry. MS/MS offers superior specificity, reduced interference, and the ability to test multiple enzymes simultaneously (e.g., in newborn screening panels).

Molecular Testing for LSD Diagnosis
Molecular testing plays a pivotal role in the diagnosis and classification of lysosomal storage disorders (LSD), complementing biochemical enzyme assays by identifying pathogenic mutations in disease-associated genes. This approach not only confirms diagnosis in cases with ambiguous enzyme activity results but also enables carrier screening, prenatal testing, and genotype-phenotype correlation, which are critical for personalized prognosis and therapy. Additionally, molecular testing helps detect pseudodeficiency alleles and variants of uncertain significance (VUS), refining diagnostic accuracy.

Both RFLP-PCR and ARMS-PCR are targeted, PCR-based techniques used in LSD diagnosis to detect known pathogenic mutations. RFLP-PCR relies on restriction enzyme digestion of PCR products to identify specific sequence variations, while ARMS-PCR uses allele-specific primers to selectively amplify mutant or wild-type sequences. However, their utility is limited to predefined variants and cannot detect novel or complex genetic alterations.

Mutation scanning techniques, such as DHPLC or HRM, screen for unknown variants by detecting heteroduplex formation or melting curve anomalies in PCR-amplified DNA. These methods are sensitive to small sequence changes and serve as a preliminary step before sequencing. While useful for variant detection, they do not identify the exact mutation and require confirmation via sequencing.

Sanger sequencing remains the gold standard for validating mutations identified through screening methods or analyzing small genes. It provides base-by-base resolution of targeted regions and is highly accurate for diagnostic confirmation and family studies. However, its low throughput and high cost per sample limit its use in large-scale or multi-gene testing.

MLPA detects exon-level deletions/duplications that are missed by sequencing, such as large rearrangements in MPS I or Gaucher disease. It uses probe hybridization and PCR amplification to quantify exon copy numbers, making it complementary to sequencing. MLPA is particularly valuable for diagnosing LSDs with CNV-prone genes but cannot detect point mutations or small indels.

NGS enables high-throughput, parallel analysis of multiple LSD-related genes, making it ideal for cases with unclear biochemical diagnoses or heterogeneous phenotypes. It identifies point mutations, indels, CNVs, and structural variants in a single assay. While NGS offers unparalleled comprehensiveness, challenges include data interpretation (VUS classification) and higher infrastructure requirements.
This concluding section synthesizes key diagnostic approaches for lysosomal storage disorders (LSD), from foundational enzyme activity assays to advanced molecular techniques. Based on the studies performed, the gold standards for enzyme activity assays and molecular diagnostics are MS/MS and NGS, respectively. While current tools have significantly improved detection rates and accuracy, challenges remain in diagnosing ultra-rare variants, reducing turnaround time, and enhancing accessibility. The future of LSD diagnostics lies in multi-omics integration, AI-driven variant interpretation, and point-of-care technologies.
At Alta DiagnoTech, we are committed to advancing the diagnosis of lysosomal storage disorders (LSDs) through innovative, high-precision IVD solutions. Our comprehensive portfolio spans enzyme activity assays and molecular genetic testing products. If you have related needs, please feel free to contact us for more information or product support.
References
| Cat.No | Product Name | Price |
|---|---|---|
| EK-YJL-2499 | Human Lysosomal Acid Lipase/cholesteryl Ester Hydrolase (LIPA) ELISA Kit | Add To Cart |
| EK-YJL-3125 | Human Lysosomal Protective Protein (CTSA) ELISA Kit | Add To Cart |
| EK-YJL-2893 | Human Lysosomal Alpha-glucosidase (GAA) ELISA Kit | Add To Cart |
| EK-YJL-2194 | Mouse Lysosomal Pro-X Carboxypeptidase (PRCP) ELISA Kit | Add To Cart |
| EK-YJL-2531 | Human Lysosomal-associated Membrane Protein 2, HLAMP-2 ELISA Kit | Add To Cart |
This article is for research use only. Do not use in any diagnostic or therapeutic application.
|
There is no product in your cart. |
Copyright © 2026 Alta DiagnoTech. All rights reserved.