Cystic fibrosis (CF) requires timely and accurate diagnosis to enable early intervention and improve patient outcomes. This resource provides healthcare professionals with an in-depth look at modern CF diagnostic approaches, including newborn screening strategies, high-sensitivity IRT assays, expanded CFTR genetic panels, and gold-standard confirmatory tests. We also explore emerging biomarkers and future directions in CF diagnostics. Whether you're establishing screening protocols or optimizing testing workflows, this guide offers evidence-based insights to enhance your CF diagnostic capabilities.
Overview of Cystic Fibrosis (CF)
Cystic fibrosis (CF) is a life-limiting genetic disorder caused by mutations in the CFTR gene that disrupt chloride and bicarbonate transport in epithelial cells. This dysfunction leads to the production of abnormally thick mucus that primarily affects the lungs and pancreas. In the lungs, the mucus causes chronic infections and progressive respiratory decline, while pancreatic involvement results in malabsorption and diabetes. The condition occurs in approximately 1 in 2,500 to 3,500 Caucasian newborns, with varying prevalence across different populations.
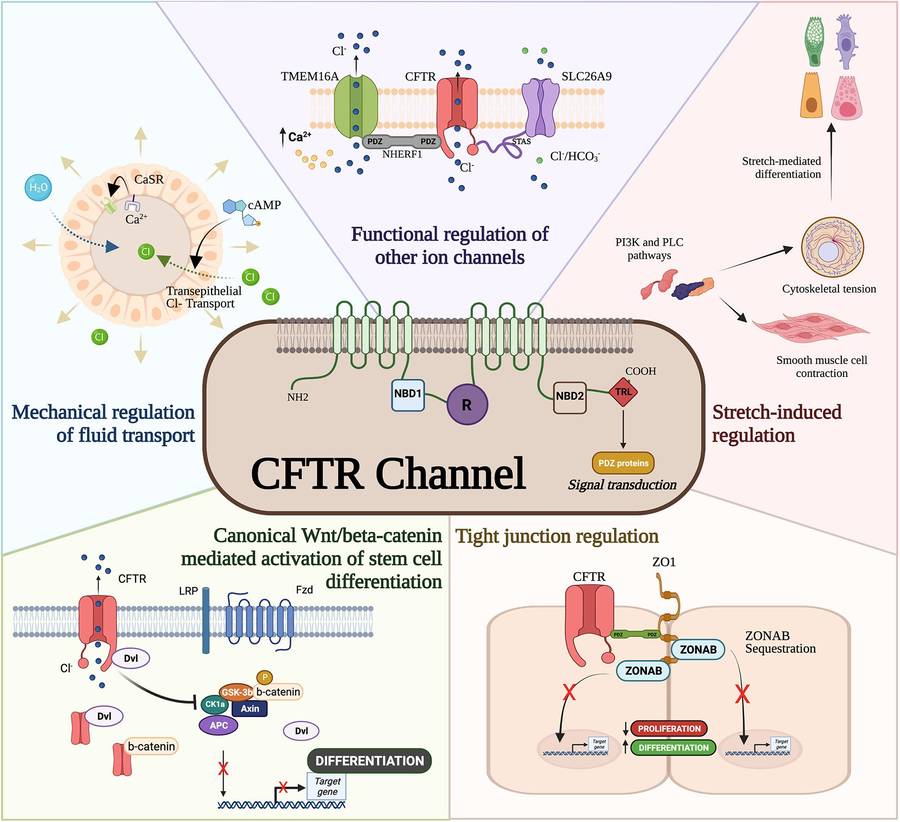 Fig.1 Cystic fibrosis (CF) transmembrane conductance regulator (CFTR) developmental interactome. (Huang E N, et al., 2021)
Fig.1 Cystic fibrosis (CF) transmembrane conductance regulator (CFTR) developmental interactome. (Huang E N, et al., 2021)
Newborn Screening for Cystic Fibrosis (CF)
Benefits of Newborn Screening
Newborn screening enables early diagnosis before symptoms develop so interventions can be initiated immediately. Studies have shown that early detection through screening can improve long-term survival, prevent complications such as malnutrition and developmental delays, and buy families more time to access specialized CF care and emerging therapies.
Challenges of Newborn Screening
CF newborn screening faces several implementation challenges. The two-step testing approach can yield false positives due to elevated IRT levels in premature or stressed infants, causing unnecessary parental anxiety. Ethnic disparities exist as standard CFTR panels may miss rare mutations in non-Caucasian populations, leading to false negatives.
Cystic fibrosis (CF) newborn screening is a critical public health initiative designed to detect affected infants shortly after birth, enabling early intervention to improve long-term health outcomes. The screening process typically follows a two-step approach: initial testing (IRT assays) and confirmatory testing (CFTR gene mutation analysis).
High-Sensitivity IRT Assays
Immunoreactive trypsinogen (IRT) assays represent the cornerstone of first-tier cystic fibrosis (CF) newborn screening worldwide. High-sensitivity IRT assays represent a significant advancement in this field, offering improved accuracy and reliability in early CF detection. This approach has the following advantages:
Enhanced Analytical Performance
High-sensitivity IRT assays significantly improve accuracy through optimized detection thresholds, enabling identification of subtle IRT elevations while minimizing false positives.
Technical Advantages
These assays support full automation, require minimal sample volume, and deliver rapid results - making them ideal for high-throughput newborn screening programs.
Clinical Benefits
The enhanced precision enables earlier intervention, reduces retesting rates, and improves screening reliability - particularly for preterm infants and diverse ethnic populations.
Expanded CFTR Panels
Expanded CFTR panels represent a significant advancement in genetic testing for cystic fibrosis (CF), offering more comprehensive mutation coverage compared to traditional screening methods. These next-generation panels utilize advanced sequencing technologies to detect a wider range of pathogenic variants in the CFTR gene, including:
- Broad Mutation Detection: Screening for hundreds of CFTR mutations beyond the standard panel
- Ethnic-Inclusive Design: Improved detection rates across diverse populations
- Variant Classification: Advanced interpretation of variants of uncertain significance (VUS)
The implementation of expanded CFTR panels in newborn screening programs and diagnostic workflows provides several key benefits:
- Higher Diagnostic Yield: Reduced false negatives through more complete mutation coverage
- Personalized Care: Better genotype-phenotype correlation for treatment planning
- Future-Ready Testing: Compatibility with evolving therapeutic options like CFTR modulators
Expanded CFTR panels serve as powerful diagnostic tools with four key clinical applications:
- Confirmatory testing after positive IRT screening
- Diagnostic evaluation of symptomatic patients
- Carrier screening in high-risk populations
- Genetic counseling for family planning
Confirmatory Testing & Beyond
Sweat Chloride Testing
The sweat chloride test maintains its position as the gold-standard confirmatory test for CF, measuring chloride concentration in sweat collected through pilocarpine iontophoresis. This non-invasive procedure provides definitive results when values exceed 60 mmol/L (positive), fall below 30 mmol/L (negative), or require clinical correlation for intermediate values (30-59 mmol/L).
Emerging Biomarkers
Emerging biomarkers are revolutionizing CF diagnostics by complementing traditional tests with novel physiological insights. Key developments include fecal elastase-1 for pancreatic insufficiency assessment, nasal potential difference measurements for direct CFTR function evaluation, and proteomic signatures like PAP/SP-D proteins for monitoring lung involvement.
While newborn screening provides initial cystic fibrosis (CF) risk assessment, confirmatory testing remains essential for definitive diagnosis and comprehensive care. The diagnostic journey extends beyond initial screening to include gold-standard validation and emerging approaches that enhance precision medicine. Two critical components shape this evolving landscape: sweat chloride testing as the diagnostic cornerstone, and novel biomarkers that may complement traditional methods.
Future of Cystic Fibrosis (CF) Diagnosis
The future of cystic fibrosis (CF) diagnosis is poised for transformative advances, driven by cutting-edge technologies and precision medicine approaches. Next-generation sequencing (NGS) will enable comprehensive CFTR profiling at lower costs, while AI-powered algorithms integrate multi-omics data for personalized diagnostic and prognostic insights. As these innovations converge, we're moving toward a new paradigm where CF diagnosis evolves from a single-point assessment to a dynamic and lifelong monitoring process.
Alta DiagnoTech provides cutting-edge
IVD solutions for cystic fibrosis (CF), featuring sweat chloride test kits, high-performance IRT assays, and customizable CFTR panels to enable comprehensive diagnosis from newborn screening to confirmatory testing. If you have related needs, please feel free to
contact us for more information or product support.
Reference
- Huang E N, Quach H, Lee J A, et al. A developmental role of the cystic fibrosis transmembrane conductance regulator in cystic fibrosis lung disease pathogenesis[J]. Frontiers in Cell and Developmental Biology, 2021, 9: 742891.
This article is for research use only. Do not use in any diagnostic or therapeutic application.




